Fonte: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Departamento de Ciências Clínicas Lund, Divisão de Medicina de Infecção, Centro Biomédico, Universidade de Lund, 221 00 Lund, Suécia
O Planeta Terra é um habitat para milhões de espécies bacterianas, cada uma das quais tem características específicas. A identificação de espécies bacterianas é amplamente utilizada na ecologia microbiana para determinar a biodiversidade de amostras ambientais e microbiologia médica para diagnosticar pacientes infectados. As bactérias podem ser classificadas usando métodos convencionais de microbiologia, como microscopia, crescimento em mídia específica, testes bioquímicos e sorológicos e ensaios de sensibilidade a antibióticos. Nas últimas décadas, os métodos de microbiologia molecular revolucionaram a identificação bacteriana. Um método popular é o sequenciamento genético RNA ribossômico 16S (rRNA). Este método não só é mais rápido e mais preciso do que os métodos convencionais, mas também permite a identificação de cepas difíceis de crescer em condições laboratoriais. Além disso, a diferenciação das cepas no nível molecular permite a discriminação entre bactérias fenotipicamente idênticas (1-4).
16S rRNA une-se a um complexo de 19 proteínas para formar uma subunidade 30S do ribossomo bacteriano (5). É codificado pelo gene 16S rRNA, que está presente e altamente conservado em todas as bactérias devido à sua função essencial no conjunto ribossomo; no entanto, também contém regiões variáveis que podem servir como impressões digitais para determinadas espécies. Essas características tornaram o gene 16S rRNA um fragmento genético ideal a ser usado na identificação, comparação e classificação filogenética de bactérias (6).
O sequenciamento genético rRNA 16S é baseado na reação em cadeia de polimerase (PCR) (7-8) seguida pelo sequenciamento de DNA (9). PCR é um método de biologia molecular usado para amplificar fragmentos específicos de DNA através de uma série de ciclos que incluem:
i) Denaturação de um modelo de DNA duplo encalhado
ii) Ressaração de primers (oligonucleotídeos curtos) que são complementares ao modelo
iii) Extensão dos primers pela enzima dna polimerase, que sintetiza uma nova cadeia de DNA
Uma visão geral esquemática do método é mostrada na Figura 1.
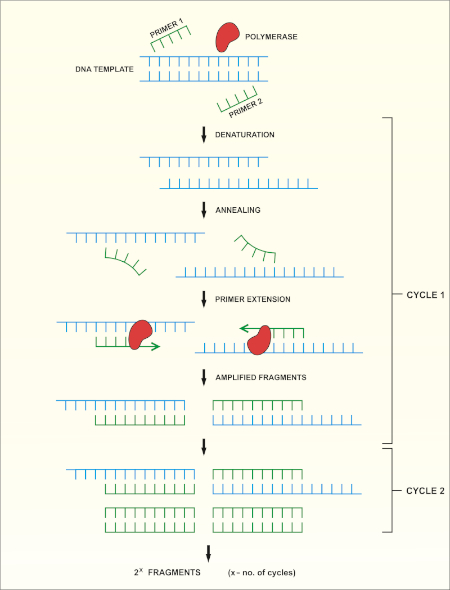
Figura 1: Visão geral esquemática da reação do PCR. Clique aqui para ver uma versão maior desta figura.
Existem vários fatores que são importantes para uma reação de sucesso do PCR, um dos quais é a qualidade do modelo de DNA. O isolamento do DNA cromossômico de bactérias pode ser realizado usando protocolos padrão ou kits comerciais. Deve-se tomar cuidado especial para obter DNA livre de contaminantes que podem inibir a reação da PCR.
Regiões conservadas do gene rRNA 16S permitem o desenho de pares de primer universais (um para a frente e um inverso) que podem se ligar e amplificar a região alvo em qualquer espécie bacteriana. A região alvo pode variar de tamanho. Enquanto alguns pares de primer podem amplificar a maior parte do gene 16S rRNA, outros amplificam apenas partes dele. Exemplos de primers comumente usados são mostrados na Tabela 1 e seus locais de vinculação são retratados na Figura 2.
| Nome do primer | Sequência (5’→3′) | Para a frente/ para trás | Referência |
| 8F b) | AGAGTTTGATCCTGGCTCAG | encaminhar | -1 |
| 27F | AGAGTTTGATCMTGGCTCAG | encaminhar | -10 |
| 515F | GTGCCAGCCCCCGGGGTAA | encaminhar | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | inverter | -12 |
| 1391R | GACGGGCGGTGTGTRCA | inverter | -11 |
| 1492R | GGTTACCTTGTTACGACTT | inverter | -11 |
Tabela 1: Exemplos de oligonucleotídeos padrão utilizados na amplificação de genes de rRNA 16S a).
a) Os comprimentos esperados do produto PCR gerados usando as diferentes combinações de primer podem ser estimados calculando-se a distância entre os locais de ligação para o primer para a frente e para o primer (ver Figura 2), por exemplo, o tamanho do produto PCR usando o par de primer 8F-1492R é de ~1500 bp, e para o par de primer 27F-911R ~900 bp.
b) também conhecido como fD1

Figura 2: Figura representativa da sequência de rRNA 16S e dos sites de vinculação de primer. As regiões conservadas são coloridas em cinza e regiões variáveis são preenchidas com linhas diagonais. Para permitir a maior resolução, o primer 8F e 1492R (nome baseado na localização na sequência de rRNA) são usados para amplificar toda a sequência, permitindo o sequenciamento de várias regiões variáveis do gene. Clique aqui para ver uma versão maior desta figura.
As condições de ciclismo para PCR (ou seja, a temperatura e o tempo necessários para que o DNA seja desnaturado, enrosco com primers e sintetizado) dependem do tipo de polimerase que é usado e das propriedades dos primers. Recomenda-se seguir as diretrizes do fabricante para uma determinada polimerase.
Após a conclusão do programa PCR, os produtos são analisados por eletroforese de gel agarose. Um PCR bem sucedido rende uma única faixa de tamanho esperado. O produto deve ser purificado antes do sequenciamento para remover primers residuais, desoxiribonucleotídeos, polimerase e tampão que estavam presentes na reação pcr. Os fragmentos de DNA purificados são geralmente enviados para sequenciamento para serviços de sequenciamento comercial; no entanto, algumas instituições realizam sequenciamento de DNA em suas próprias instalações principais.
A sequência de DNA é gerada automaticamente a partir de um cromatgrama de DNA por um computador e deve ser cuidadosamente verificada para a qualidade, pois a edição manual às vezes é necessária. Após esta etapa, a sequência genética é comparada com sequências depositadas no banco de dados de rRNA 16S. As regiões de semelhança são identificadas, e as sequências mais semelhantes são entregues.