Organelle morphology and dynamics are subject to change as yeast cells respond to external and internal signals. Here, we provide representative images of yeast organelles in the mid-log phase (Figure 3A,B). As mentioned previously, several organelles have their distinct morphological features, thus are easy to recognize without extensive comparison with other organelle markers. These include ER, mitochondria, and vacuoles. Note that in some laboratory strains, including the ones we are showing here, the fission-fusion balance of vacuoles is skewed to the extent that many log-phase cells contain a single vacuole. Golgi apparatuses, endosomes, lipid droplets, and peroxisomes are generally too small for normal light microscopy to resolve their contours. So, they all appear as cytoplasmic dots. Ascertaining their identities requires cross-comparison with other organelle markers, which we have performed for the present marker set in log-phase cells7.
As presented in the Introduction, most of our marker constructs have the potential to be integrated multiple times in the genome. To avoid unnecessary complications, transformants with single-copy integration can be screened when verifying their marker expression by fluorescent microscopy (Figure 2B). The fluorescence signal in transformants carrying multiple copies is much brighter than those with a single copy.
Preparation of microscope slides containing live yeast cells is a simple procedure that can be done quickly to screen through tens of samples in a single experiment. However, it takes some practice to find the "right" pressure that secures a single layer of cells on slides without crushing them (Figure 1B). Too little force, cells stay in multiple layers and likely float around; too much force, the cells are essentially mechanically lysed. With experience, one can also spot dead cells and old cells, which should rarely appear in wild-type log-phase populations.
Time-lapse imaging is a powerful technique for investigating membrane dynamics. Here we demonstrate the frequent formation and consumption of autophagosomes in starved yeast cells labeled with GFP-Atg8 (Figure 4). There are claims in the literature that a yeast cell only has a single subcellular site that generates autophagosomes. It sounds correct if one only checks single-time-point snapshots since, on average, there is about one dot per cell. However, one can see easily in time-lapse imaging that in a single yeast cell, different instances of autophagosome biogenesis can occur independently at different subcellular locations over time.
Besides tracking the dynamics of each organelle, another common use of organelle markers is to determine the subcellular localization of a protein of interest. In practice, proteins in cells are often localized to more than one organelle. Here, we use Sft1 as an example; Sft1 is a v-SNARE functioning in intracellular transport13. GFP-tagged Sft1 manifests as multiple cytoplasmic dots (Figure 5). By cross-comparison with co-expressed red organelle makers, one can see that Sft1 partially colocalizes with early Golgi and late Golgi/early endosome markers, which provide important clues for understanding Sft1 function.
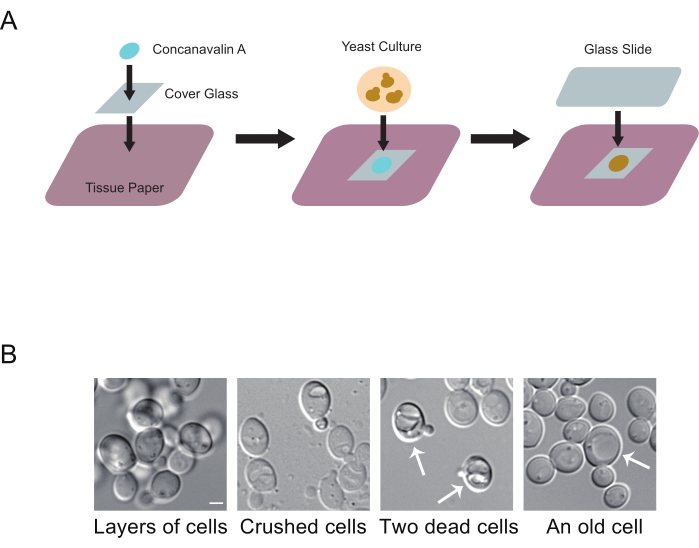
Figure 1: Sample slide preparation. (A) Carton illustration of the preparation. (1) Place a cover glass on tissue paper, spread the concanavalin A solution to the top side of the cover glass, and wait for 5 min or longer. (2) Add ~100 µL of yeast culture medium to the top side of the cover glass, and wait for 5 min for the cells to settle. (3) Cover the cover glass with a glass slide and press to combine it with the cover glass. Excess liquid is absorbed by the underlying tissue paper. (B) Appearance of yeast cells in undesirable situations. From left to right: formation of layers of cells when insufficient force is applied in slide preparation; crushed yeast cells when excessive force is applied in slide preparation; two potentially dead cells, marked by arrows; an old cell with a very large vacuole, marked by arrows. Scale bar: 2 µm. Please click here to view a larger version of this figure.
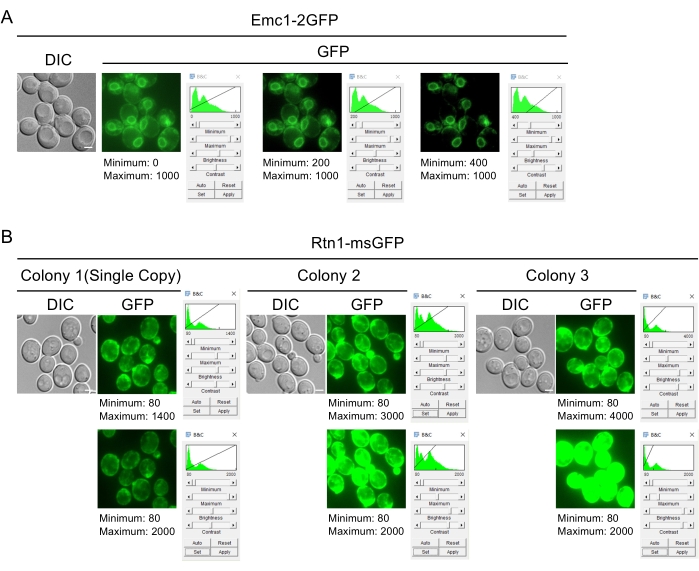
Figure 2: Adjusting image display and identifying single-copy integration transformants. (A) Effect of adjusting minimum display value. The visual appearance of a 16-bit image in ImageJ/Fiji depends on the user-determined display range. Here, the effect of adjusting the minimum value on the appearance of the endoplasmic reticulum is shown. The value can be set in the Brightness and Contrast (B&C) window. The visual appearance of the same 16-bit image varies as this setting is changed. On the left, with a value of 0, the contrast is low, with the background signal visible in areas devoid of cells. On the right, a value of 400 is substantially higher than the background; as a result, the peripheral endoplasmic reticulum (i.e., the network beneath the plasma membrane) becomes obscured. The histogram in the Brightness and Contrast (B&C) window provides information on the distribution of signal intensity across the image, which can be used as a guide in picking the appropriate display range. Scale bar: 2 µm. (B) Inferring construct integration number among transformants by comparing the fluorescent signal intensity. Note that the same exposure parameters should be employed when capturing the images. In this example, images from three different transformants are being compared. Colony 1 is a single-copy-integration transformant; colony 2 and colony 3 are multi-copy-integration transformants. In the top row, different minimum and maximum thresholds were applied to each image (80-1400, 80-3000, 80-4000, respectively), with higher maximum values applied to brighter samples. In this view, similar subcellular distribution patterns can be seen in all three samples. In the bottom row, the same minimum and maximum thresholds (80-2000) were applied to all three images. In this view, higher signal levels in colony 2 and colony 3 are apparent, reflecting the fact that they carry multiple copies of the Rtn1-msGFP construct in their genomes. Scale bar: 2 µm. Please click here to view a larger version of this figure.

Figure 3: Typical morphology of major yeast organelles. (A) Representative snapshot images. The fluorescent protein constructs and the organelles they represent are indicated to the left of the images. DIC: differential interference contrast, a single slice. Slice: A single slice in the z-stack of the fluorescent channel. Projection: Max intensity projection of the fluorescent channel. Note that here the mapping of 16-bit to 8-bit was done differently for slices and projections to enhance the visibility of organelles in slices. Scale bar: 2 µm. (B) Morphological differences between nuclear endoplasmic reticulum/nuclear envelope and vacuole. Both the nuclear endoplasmic reticulum and vacuoles manifest as circular structures when imaged in the center of the cell. Compared with the vacuoles, the nuclear endoplasmic reticulum is generally less rounded, and the corresponding area in DIC is also less pronounced. Scale bar: 2 µm. Please click here to view a larger version of this figure.
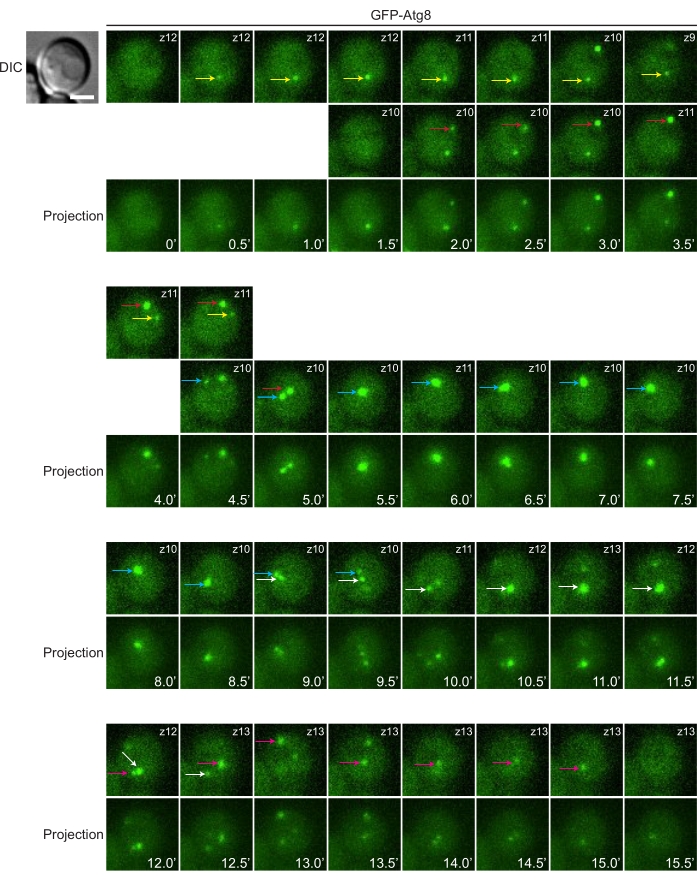
Figure 4: Following the dynamics of autophagosomes by time-lapse imaging. Yeast cells expressing GFP-Atg8 were grown to mid-log phase in YPD medium, then shifted to SD-N medium for starvation. After 45 min of the medium switch, yeast cells were mounted on glass-bottom dishes and incubated with SD-N for time-lapse imaging. Image stacks were collected at 30 s intervals. In this example, five rounds of autophagosome formation and disappearance can be seen. The dots, representing Atg8-positive autophagic structures, were manually tracked over time, and labeled with arrows of different color (i.e., arrows of the same color denote the same dot being followed). Note that at any particular time point, different dots (if there is more than one) may appear in different z-positions. Individual slices containing the tracked dots in focus are shown in the top one or two rows, with the z-position indicated in the top right corner (i.e., z10 denotes the 10th slice, and z12 denotes the 12th slice). The use of max-intensity projection is a convenient approach to glance through all structures. However, the contour of each individual structure is not as clear as in single slices. Max intensity projections are shown in the row below the slices. Scale bar: 2 µm. Please click here to view a larger version of this figure.
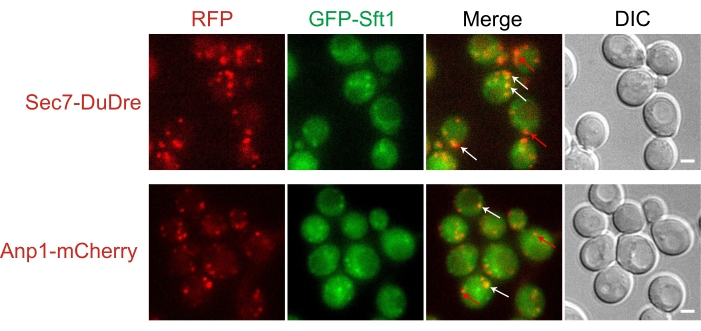
Figure 5: Using organelle markers to determine protein subcellular localization. Representative slices of yeast cells co-expressing GFP-Sft1 with either Sec2-DuDre (late Golgi/early endosome marker) or Anp1-mCherry (early Golgi marker). Sft1 displayed partial colocalization with both organelle markers. White arrows, the incidence of colocalization; red arrows, the incidence of no colocalization. Scale bar: 2 µm. Please click here to view a larger version of this figure.
Supplementary Figure 1: Screenshots showing the selection of imaging parameters in VisiView software. (A) Setting excitation light intensity. As a starting point, set 100% for all channels. See text for general considerations in adjusting excitation parameters. (B) Selecting the light channels to be imaged. This example demonstrated the selection of two fluorescence channels: GFP and mCherry. In VisiView, each channel needs to be picked from a drop-down menu. In many other software applications, all channels can be configured in a spreadsheet-like single graphical interface. Note that in this particular software, there is a checkbox Same Exposure/Gain for all Wavelengths, which needs to be unchecked to allow unique exposure duration for each channel. (C) Setting the number of slices and stepping for collecting z-stacks. 3.5 µm in each direction, covering 7 µm of depth at 0.5 µm steps. (D) Picking the order of channel switching and z-stack progression in multichannel imaging. In most microscope controlling software, one can choose to finish all channels at a z-position before moving on to the next slice, or finish a complete z-stack in each channel before moving to the next channel. This choice influences both time consumption in image acquisition and accuracy of colocalization for moving objects. (E) Setting the number of time points and intervals for time-lapse imaging. Please click here to download this File.
Table 1: List of organelle marker plasmids. Please click here to download this Table.
