Whole-mount 3D confocal/2-photon microscopy reconstructions of the sternal bone marrow time course revealed the engraftment and expansion of transplanted co-5FPs in a pattern with remarkable characteristics: clones appeared clearly delineated, homogenously marked with wide palette of colors initially and progress over time to preferentially contain cells of mostly one color. Confocal microscopy setup and representative examples of imaging 5FPs-marked HSPC in the sternal bone marrow are illustrated in Figure 2 and Movies 1 and 2.
Whole-mount 3D confocal/2-photon microscopy reconstructions of intact tissues demonstrate the possibility to trace the fate of color marked BM-derived cells for extended time periods in the live tissues. Representative examples of bone-marrow derived cells in hematopoietic and non-hematopoietic organs following transplantation are shown in Figure 3.
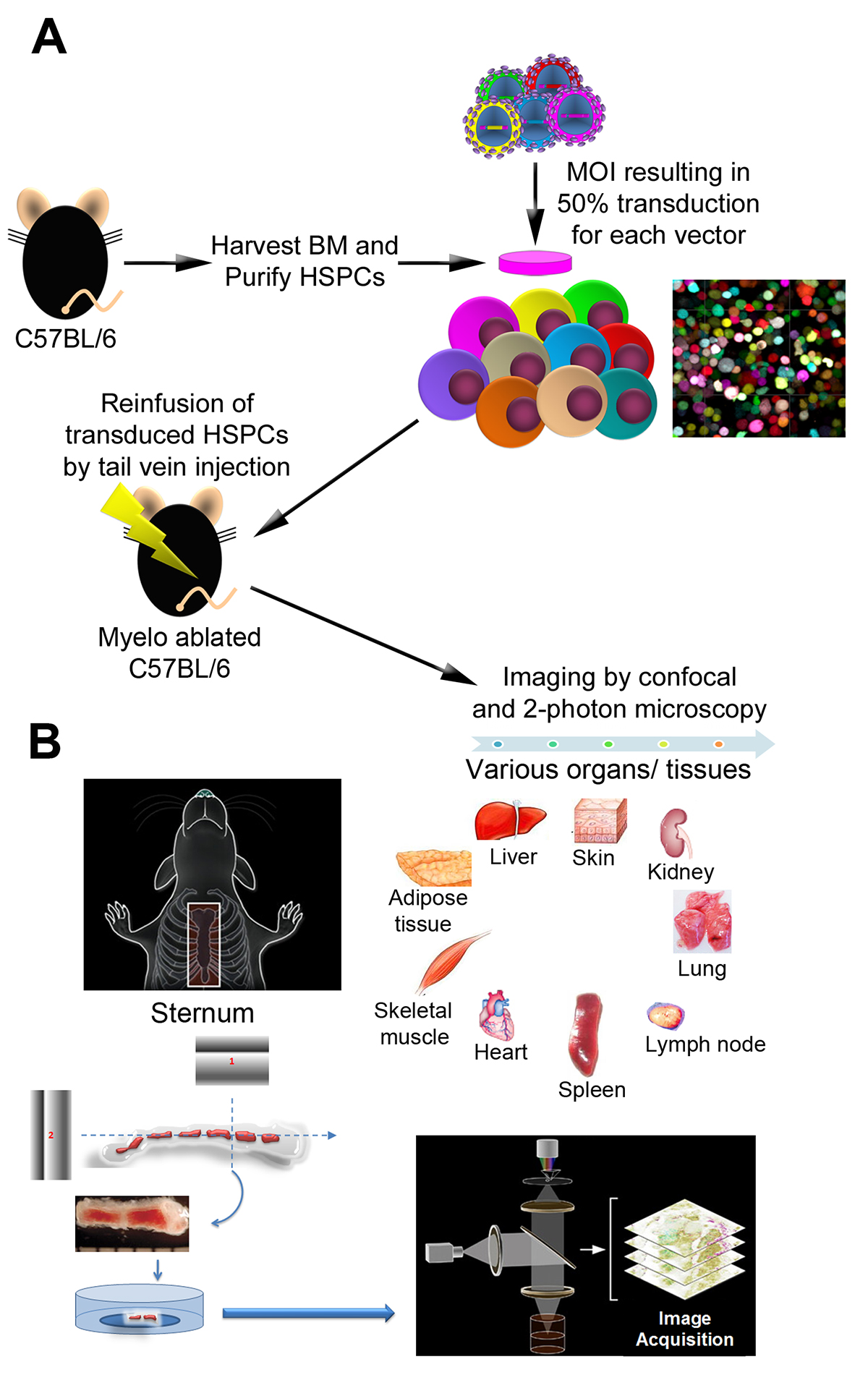
Figure 1. Overview of the experimental procedures. A) The schematic steps illustrate isolation of Lin- BM cells, transduction with LeGO vectors encoding a variety of FPs color variants, and reinfusion of transduced cells into-myelo-ablated mice (bone marrow transplant). B) Bone marrow (sternum, calvaria), as well as various organs/tissues were examined by confocal and two-photon microscopy at different time points following transplant. Panel A is adapted from Figure 1 in Malide et al.4 and Panel B adapted from Figure 1 in Takaku et al3.
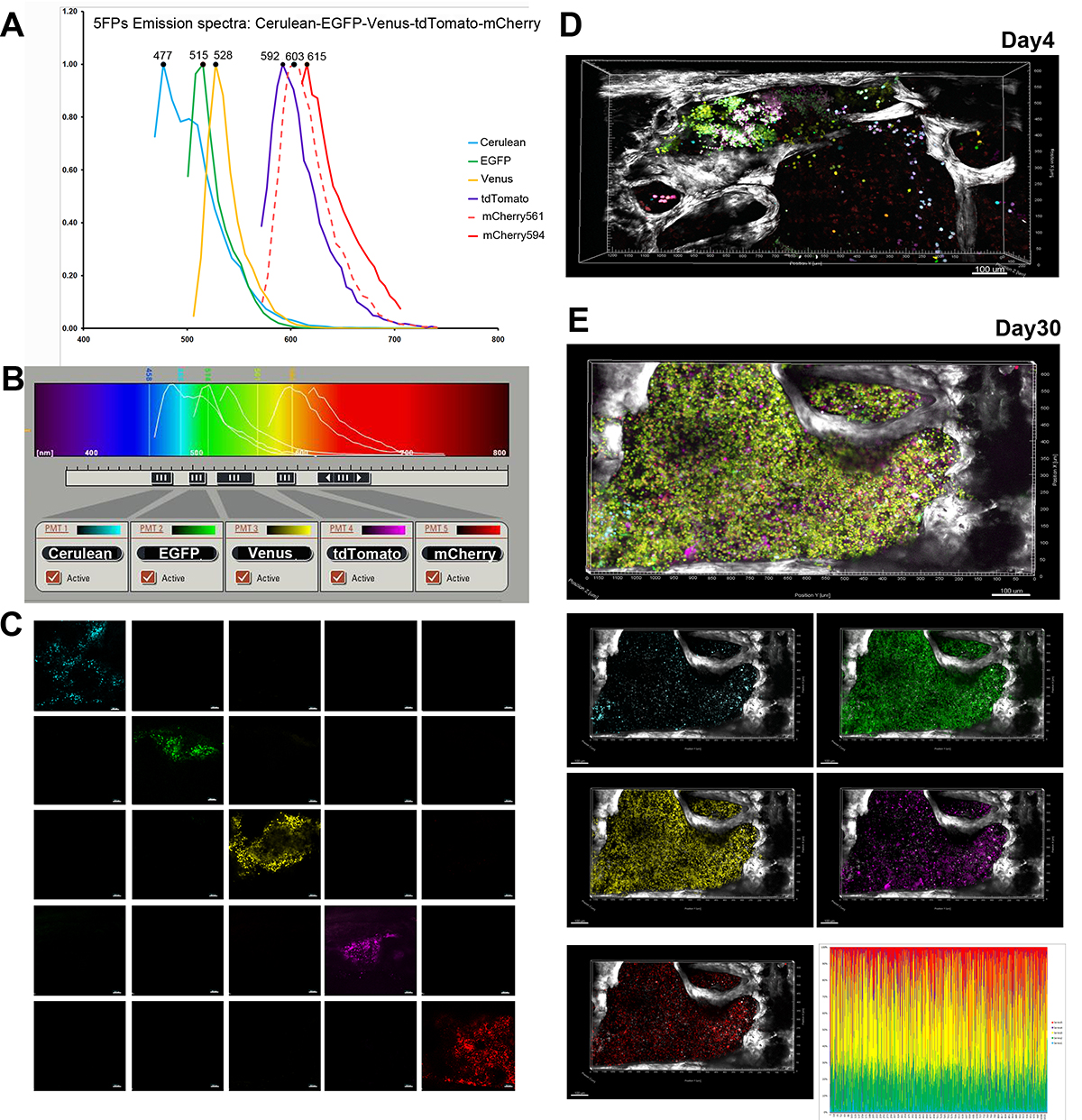
Figure 2. Confocal microscopy setup and representative example for imaging 5FPs-marked HSPC in the bone marrow. A) Spectral (xyλ) imaging used to record reference emission spectra for each FP, illustrated in normalized histograms pseudocolored with cyan (Cerulean), green (EGFP), yellow (Venus), magenta (tdTomato), and red (mCherry): excited by 594 nm- (solid red line) compared to 561 nm- (dashed red line) wavelength. B) Five channels set to image sequentially emission of: Cerulean (468-482 nm), EGFP (496-514 nm), Venus (523-558 nm), tdTomato (579-597 nm) and mCherry (618-670 nm). Panels A and B are adapted from Figure 1 in Malide et al.4 C) Imaging of sternal BM from mice transplanted with transduced with individual FP vectors. Each FP was visible only in the cells transduced with the corresponding vector imaged in the appropriate channel, and absent from the others (no cross-talk). D,E) Engraftment and expansion of transplanted co-5FP marked cells in the bone marrow in vivo. At day 4 post-transplant (D) clusters of cells marked in wide variety of colors were visible close proximity to the bone edge (SHG, white), preferentially located adjacent to the joint between two fossae. By day 30 (E) one large clone of unique color (green – yellow) has expanded to occupy the entire fossae as illustrated in the merged as well as single channels images. FPs content analysis demonstrates homogeneous marking by 2 FPs variants EGFP and Venus.
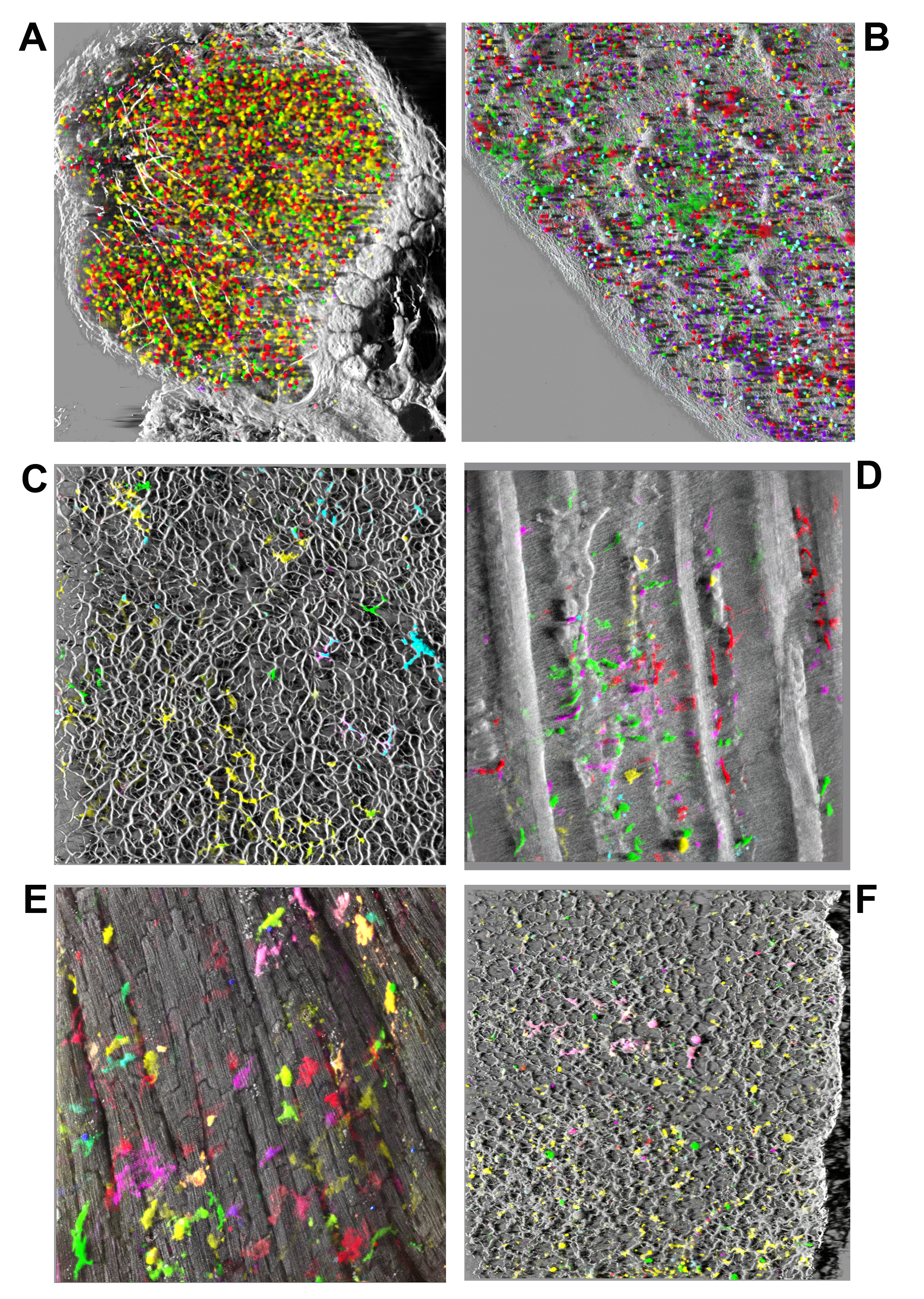
Figure 3. Examples of bone-marrow derived cells in hematopoietic and non-hematopoietic organs following transplantation. Various tissues and organs were imaged intact through depths of 150-200 µm and large surface areas, computationally stitched and rendered in 3D as shadow reconstructions. A) Popliteal lymph node at 120 days post-transplant demonstrates scattered cells mostly marked in yellow (Venus) and red (mCherry) peripherally surrounded by collagen fibers (white, SHG). B) Similarly, the spleen at the same time, displays small clusters and mostly scattered cells of various colors intertwined by collagen fibers network. C) In the liver fluorescent cells with morphology suggestive of stellate cells, or macrophages (Kupffer cells) of various colors were aligned along collagen fibers network (SHG white) delineating hepatic lobular structures. D) In an skin flap imaged from dermal side fluorescent cells of diverse colors and morphologies were seen, most with large size and morphology suggesting Langerhan’s or dendritic-like identity, lying under elastin fibers (autofluorescence at 780 nm, white) and along collagen (SHG at 920 nm, white) muscle fibers and hair follicles. E) In the heart viewed from the epicardial side numerous individual large fluorescent cells with macrophage-like morphology originating from multiple clones based on the diverse colors seen are visible superficially and also interspersed deeper between cardiomyocytes (white) visualized by their intrinsic two-photon autofluorescence (at 780 nm). No FP fluorescent cardiomyocytes were observed. Nearby cells (C-E) of the same color suggest in situ proliferation and short-distance migration. F) In the lung numerous fluorescent cells with a diverse palette of colors were scattered throughout the lace-like structure of collagen fibers (SHG) at all depth levels, with variable morphologies suggesting dendritic cell, macrophage, and type 2 pneumocyte identities.
Movie 1. 3D reconstruction of sternal BM at 4 days post-transplant Co 5FPs. Clusters of cells marked in wide variety of colors were visible in close proximity to the bone edge (SHG, white), preferentially located adjacent to the joint between two fossae. Please click here to view this movie.
Movie 2. 4D time lapse sternal BM two-photon and OPO microscopy at day 39 post transplant Co 3FPs (Cerulean – white, Venus – yellow, tdTomato – magenta) bone (SHG in white). Note several static clones marked in yellow or in yellow-magenta combination adjacent to bone in contrast to highly motile individual myeloid progenitor cells. Please click here to view this movie.
