Since viral replication compartments (RC) are subnuclear viral-induced structures composed of proteins and nucleic acids, similar to other nuclear domains, they proved to be amenable to isolation by velocity gradients based on biochemical features. Critical steps in the fractionation protocol are illustrated in Figure 1. At each step the samples need to be monitored by bright field microscopy to ensure integrity of the different sub-cellular fractions. For example, when swelling the cells, incubation time in the hypotonic buffer needs to be standardized in order to swell the cytoplasm avoiding damage to the nuclei. After cell homogenization, intact nuclei, free of cytoplasmic components including endoplasmic reticulum membranes, need to be obtained. Also, sonication time needs to be standardized in order to rupture the nuclear membrane of all cells without disrupting RC.
After obtaining the sub-nuclear fractions, key controls need to be included to determine the association and enrichment of bona fide RC markers in the RCf. Adenoviral RC are commonly visualized in infected cells by immunofluorescence using antibodies against the viral E2A-72K protein (DBP). DBP is a viral protein that participates directly in viral genome replication; therefore, the presence of DBP in particles enriched in the RCf demonstrates the direct association of this viral protein with the isolated particles, as shown in Figure 1. Furthermore, detection of DBP in RCf by Western Blot confirms the association of this protein to the isolated RC. In Figure 2 it is shown that by late times post-infection (36 hpi), DBP is enriched in the RCf compared to the nucleoplasmic fraction (Npl), demonstrating that RC obtained with this procedure at different times post-infection reflect the expected temporal pattern of DBP association to RC. An essential viral component of RC is the viral DNA itself. In experiments like that shown in Figure 3 we demonstrate that increasing amounts of viral DNA associate with RCf as the replication cycle of the virus progresses, indicating that, as for DBP, the temporal pattern of DNA replication in these fractions can also be studied.
Besides containing viral proteins and the viral genome, RC are also sites of viral late gene expression. In Figure 4 we present representative results of experiments designed to measure by RT-PCR the level of various species of viral late mRNA and their segregation between RCf and Npl at 36 hpi. Viral late mRNA are synthesized in RC and their postranscriptional processing initiates in these sites; later these viral mRNA dissociate from RC, are liberated to the nucleoplasm, and are subsequently exported to the cytoplasm. The total pool of mature viral late mRNA (ML mRNA) was measured using primers that amplify an exon junction of the tripartite leader, a sequence that constitutes the 5’ of all adenoviral late mRNA (Figure 4A). Exon junctions in the mRNA of specific mature transcripts of the L2, L4 and L5 families were also measured. It has been established that as the late phase of the viral replication cycle progresses, increasing amounts of viral late mRNA are exported to the cytoplasm; however, by late times production of the L5 mRNA species increases further in comparison with the other late mRNA families. In the representative results sown in Figure 4 an approximately 2.5-fold difference in ML mRNA is observed in Npl compared with RCf, as expected. Interestingly when we compare mRNA from specific families, both L2 and L4 mRNA appear to be distributed in similar levels between both subnuclear fractions (Figure 4B and C, respectively), while in contrast, the L5 mRNA showed an almost 2-fold increase in Npl compared to RCf. These results suggest a differential pattern in the synthesis and liberation of the different viral late mRNA species from adenoviral RC (as has been suggested before10). Significantly, these results also demonstrate that precise measurements of the different steps in the biogenesis of the viral mRNA can be performed using isolated RC with this novel procedure.
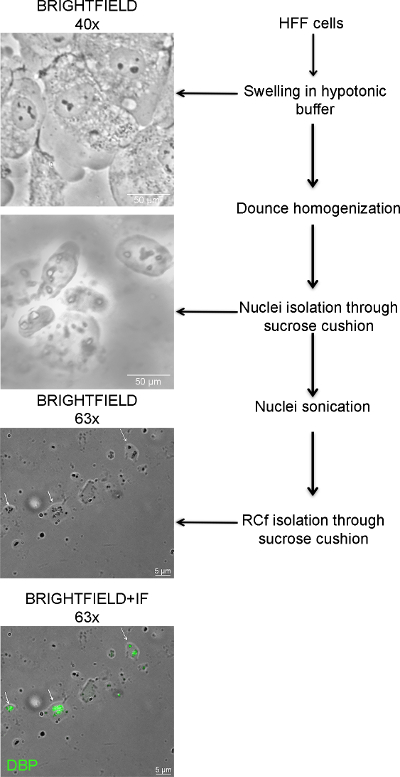
Figure 1. Bright Field Microscopy of Different Steps during the Fractionation Procedure. During the isolation of RC, the samples need to be monitored by an optical microscope to ensure integrity of sub-cellular fractions. The figure shows the steps used in the procedure to obtain RCf, from the swelling of HFF cells, to separation of nuclei and isolation of RCf through sucrose cushions. 40x micrographs: scale bar 50 µm; 63x micrographs: scale bar 5 µm; DBP is shown in green. Please click here to view a larger version of this figure.
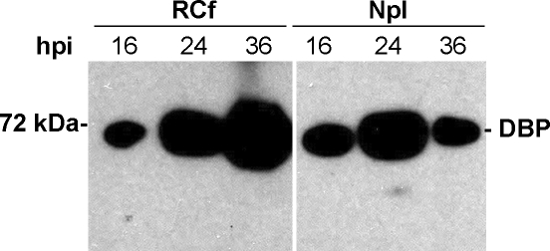
Figure 2. Western Blot against DBP. The samples are analyzed by SDS-PAGE and processed for western blot against DBP, a bona fide marker of viral RC. To determine the enrichment of the protein in RCf, it is useful to compare the presence and relative abundance of DBP in both RCf and Npl at different times post-infection. In HFF cells 16 hpi represents an early time during the adenoviral replication cycle; 24 hpi marks the transition to the late phase of infection as viral DNA synthesis begins; 36 hpi represents a late time post-infection. The expected molecular mass of DBP is shown. Please click here to view a larger version of this figure.
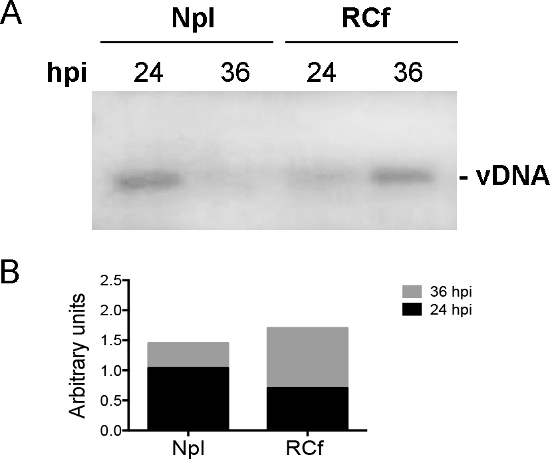
Figure 3. PCR Assay to Detect Viral DNA (vDNA) in RCf. DNA was purified from RCf and Npl at 24 and 36 hpi. Viral DNA was amplified by PCR using primers specific for the viral genome. The graph shows the enrichment of viral DNA in RCf at 36 hpi. Please click here to view a larger version of this figure.
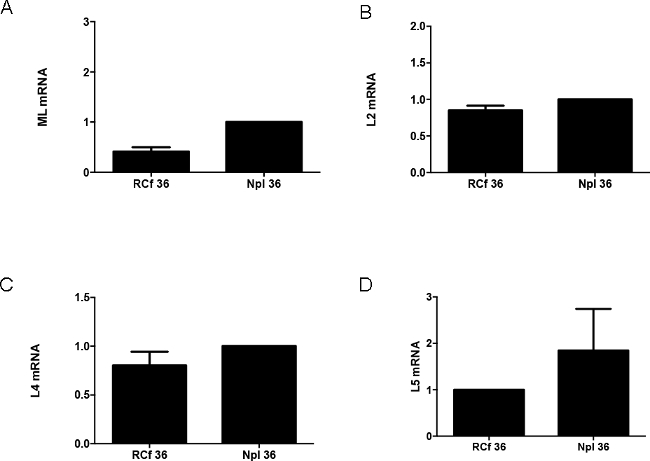
Figure 4. Analysis of Viral Late mRNA. RNA was isolated from RCf and Npl and analyzed by RT-PCR to detect specific viral late mRNA. (A) Total Viral Late mRNA (ML: Major Late); (B) mRNA from the L2 Family; (C) mRNA from the L4 Family; (D) mRNA from the L5 Family. Values represent mean ± standard deviation of triplicate samples. Please click here to view a larger version of this figure.
| Name | Forward primer sequence (5'-3') | Reverse primer sequence (5'-3') | Annealing temperature |
| ML mRNA | GCCTCCGAACGGTACTCCGCC | CGCCACGGTGCTCAGCCTACC | 60 ºC |
| L2 mRNA | GTCACAGTCGCAAGATGTCCAAGC | GCAACGCCAGCATGTCCTTATGC | 58 ºC |
| L4 mRNA | CCTCCGAACGGTACTCCGC | CCTTGCTCATCTTGCGACTGTG | 58 ºC |
| L5 mRNA | GTCACAGTCGCAAGATGAAGCG | GGTAACTAGAGGTTCGGATAGGCG | 60 ºC |
Table 1. Primers Used for Viral Late mRNA Amplification. ML mRNA: these primers allow the amplification of a region within the tripartite leader, the 5’ sequence that is common to all viral late mRNA; L2 mRNA primers allow the amplification of a specific region within the pV mRNA; L4 mRNA primers allow the amplification of a specific region within the 100 K mRNA; L5 mRNA allows the amplification of a region within the fiber mRNA. The sequence and annealing temperatures for each primer are shown.
