Cell Concentration Calculator
Figure 1 presents the overall process of CCC calibration and countable image acquisition. Figure 1A and 1B depict the P-square calibration image and calculation of P-square length in pixels. CCC determines cell concentration in a given volume using the formula:
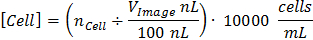
A hemocytometer's P-square has a volume of 100 nL (1 mm x 1 mm x 0.1 mm) and given this constant, the total image volume can be calculated after converting pixels to mm. Figure 1C is an ideal countable image with in-focus cells displaying the characteristic phase illumination and no background hemocytometer graduations. Finally, the scatter plot of Figure 1D shows a highly correlated, manual versus automated count of cells from 57 images taken over various experiments and cell types including HTR8, ES-2, and Swan71. Of note, the upper range of concentration was ~5.6 x 106 cells/mL with a low end of ~2.3 x 103 cells/mL (≈ 5 cells per image). It is suggested that if counts are below 10-15 cells per image, the sample should be suspended in a smaller volume to increase statistical power.
Acquisition and Processing of Images for Migration Assay Counter
Hundreds of migration assay membranes were imaged over many experiments, all of which were seeded with the Human trophoblast cell line HTR8, Swan71, or ovarian cancer cell line ES-2. Of these images, multiple were chosen to represent a range of categories from very poor to excellent quality based on brightness, clarity and color of stained cells, and the degree of background staining and unwanted particles (noise). Using these images, the default RGB Threshold color settings (≈ 150, 120, 0) were determined (Figure 2C) and used as a baseline in all subsequent developments of the algorithm. The goal was to maximize the nuclear color to total color ratio, i.e., the majority of colored pixels should be within cell nuclei. The upper panel in Figure 2A depicts the ideal image brightness, cell nuclei clarity and color, and negligible background noise. The brightness of the image is important to ensure there is a great enough contrast between cell and background to produce a black and white binary image.
If this difference is not met, large or unpredictable areas may be counted by the ImageJ Analyze Particles function; the best case scenario is a completely white background. In opposition, the lower panel of Figure 2A has extreme background staining and cells that are nearly indistinguishable. Membranes with this degree of staining will most likely produce unreliable results from the migration assay counter.
In some instances, increasing brightness to the ideal level may affect image fidelity by overexposing the image. Similarly, high exposure or brightness may produce systemic chromatic effects with progressive or irregular light and dark regions. With flatfield correction, these effects can be minimized or removed entirely as shown in Figure 2B (upper vs. lower panel). Furthermore, flatfield correction is a good way to equalize the brightness of multiple membrane images.
Calibration and Validation of Migration Assay Counter
To assist the user in better determining if migration assay membrane images meet the required criteria for accurate counting, two qualifiers were designed, called image quality (Q), and calibration recommendation (CR). Importantly, both qualifiers, as the name CR suggests, are recommendations only and act as guides rather than absolute judges of the countability of each image. Both Q and CR are based on the metrics of the frequency scatter plot of particle area (10.3.2). For simplification, a metric can be regarded as the various shapes of the curve that make up the frequency plot. The desired metric of an adequate Q (≥0.5) was determined by the observed approximate normal distribution of cell size. Commonly, cells that are unresolvable from each other, over-stained, or just particularly large, shift the skewedness to a right-tailed normal distribution (Figure 3B). As such, this is the ideal metric of a typical calibrated image. With the addition of background noise, this normally leads to a large number of particles in the 1-5 pixel area range (Figure 3A). In order to calculate the plot metrics, the data are fitted with ten Savitzky-Golay smoothed curves of differing polynomial degrees produced by Dr. Michael Thomas Flanagan's Java Scientific Library (http://www.ee.ucl.ac.uk/~mflanaga/java/). Essentially, this creates multiple points in the approximate region of each extremum. Through a series of density cluster maps of local minima and maxima, a general picture of the plot metrics can be computed as an ordered list, i.e., minimum, maximum, minimum/maximum overlap, …, nth extremum. In brief, the degree to which the smoothed curves fit the frequency data determines how tightly packed the extrema points are. The greater the clustering of non-overlapping extrema, the greater Q becomes. In theory, Q represents overall image clarity based on the distribution of distinct particle sizes.
Whether the Boolean CR is true or not depends on the sequence of extrema. From Figure 3A, the ordered list would be minimum, maximum, and a sequence of overlapping extrema based on the properties of the Savitzky-Golay curve. Since Figure 3A represents an uncalibrated image, this general sequence of extrema flags CR as true, suggesting to the user that the image may need calibration. Moving forward, it can be seen that from Figure 3B, this list would be identical but with the exclusion of the first minimum. Thus the metrics of ideal uncalibrated and calibrated images were determined. Deviations from these metrics will generally flag an image for calibration and reduce Q ≤ 0, such as the case with the high background noise membrane in Figure 3C. Applying these qualifiers to a selection of modest to excellent images, with regards to brightness and background noise, it is clear that calibrated image counts are significantly closer to manual counts than uncalibrated (1.9% ± 0.3 vs. 21.7% ± 2.9, respectively; Figure 3D, E). Given the overall high quality of images chosen for analysis (uncalibrated  = 0.71 ± 0.04; mean ± SE), there was an expected modest increase only in Q following calibration ( = 0.90 ± 0.04). Taken together, Q suggests the overall countability potential of an image whereas CR is a strong indicator of whether calibration is successful or not.
= 0.71 ± 0.04; mean ± SE), there was an expected modest increase only in Q following calibration ( = 0.90 ± 0.04). Taken together, Q suggests the overall countability potential of an image whereas CR is a strong indicator of whether calibration is successful or not.
Timing Comparisons of Both Cell Concentration Counter and Migration Assay Counter
Two researchers (User 1 and User 2) were used to test the speed comparisons between manual and automated methods. Both researchers had substantial experience with cell counting and the migration assay but User 1 was experienced in the usage of both CCC and TC while User 2 had to follow written instructions. Figure 4A compares CCC calibration time between User 1 and 2. As expected, User 1 was substantially faster than User 2, together, taking on average approximately five minutes for CCC calibration. In order to compare manual hemocytometer counting and automated rates, the manual rate of counting using a tally counter was compared to the time it took to take nine images of the hemocytometer chamber and counted in CCC (Figure 4B). A typical cell concentration of 1.15 x 106 cells/mL was used to compare timings, leading to an average of 1x increase in throughput. This rate will vary depending on the number of cells loaded into the hemocytometer as the total time taken to capture images and process them is independent of cell number.
Lastly, timings encompassing image acquisition of membranes, quantification within TC, and manual adjustments of these images was measured in Figure 4C. Notably, 12 migration assay membranes with low cell number (Total = 10,571 cells) and substantial background staining and cellular debris were chosen to facilitate a worst case scenario that would require manual adjustments to cell number. This is reflected in Figure 4C adjustment (Adj) column where it took an average of 13 min to remove unwanted counts and add missed cells. For comparison to manual counting, optimal cell counting rates were determined with Cell Counter; high quality membrane images with high cell density were used (results not shown). This yielded an average maximal rate between User 1 and 2 of 9.1 x 103 cells/h (~2.5 cells/s). Using these numbers, the membranes from Figure 4C were counted 4.4x faster with TC than would be expected at maximal manual rate. The time savings are directly dependent on cell number and image quality, by counting migration membranes that required little or no adjustment and high cell density (~7,000 cells/membrane), TC generated cell counts 1,395x faster than the maximal manual rate.
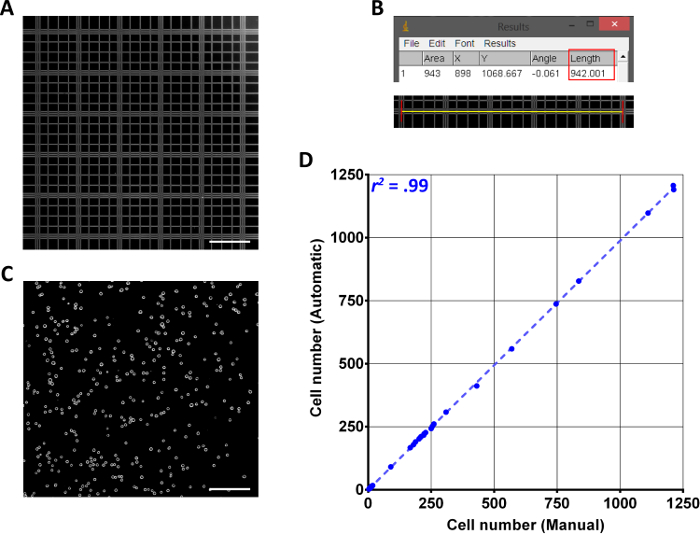
Figure 1: Cell Concentration Calculator. (A) Phase contrast micrograph of the central primary square of the hemocytometer taken at 40X total magnification. Scale bar represents 200 µm. (B) A cropped version of the image from (A) depicting what length to measure on the hemocytometer. The Results window Length column was highlighted for easy identification. Yellow bar = 1 mm. (C) An ideal micrograph of a hemocytometer containing HTR8 cells after microscope and software calibration at 40X total magnification with a resolution of 1,600 x 1,200 px. Scale bar = 200 µm. (D) A scatter plot comparing manual counts using the ImageJ plugin Cell Counter compared to automated counts of the same images using Cell Concentration Calculator. n = 57 images. Please click here to view a larger version of this figure.
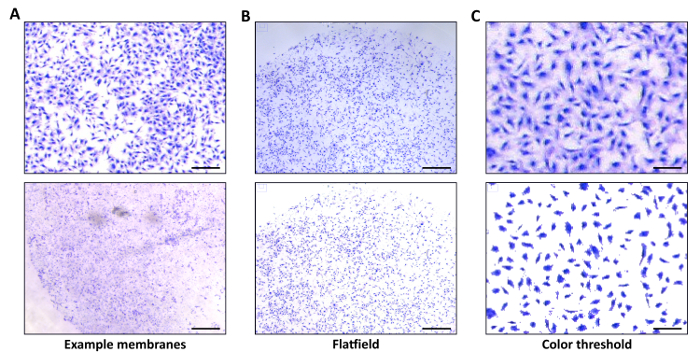
Figure 2: Migration assay counter representative images. (A) The upper panel is a one eighth portion of a total membrane depicting an ideal image with minimal background; Scale bar = 200 µm. The lower panel contains an image with significant background staining that severely affects the accuracy of the automatic counting; Scale bar = 585 µm. (B) The upper panel represents a dark image of good quality which produced inaccurate counts. The bottom panel is a countable version of the same image after flatfield correction. Scale bars = 593 µm. (C) The upper panel represents a zoomed-in view of a typical membrane. The lower panel is the same image followed by the desired ImageJ color thresholding (RGB Threshold = 150, 120, 0) to remove intercellular background and the majority of the visibly stained cytoplasm. All images were taken at 1.35X total magnification with a calibrated dissecting scope at a resolution of 2,592 x 1,944 px from multiple migration assay invasions of HTR8 stained with hematoxylin. Scale bars = 100 µm. Please click here to view a larger version of this figure.
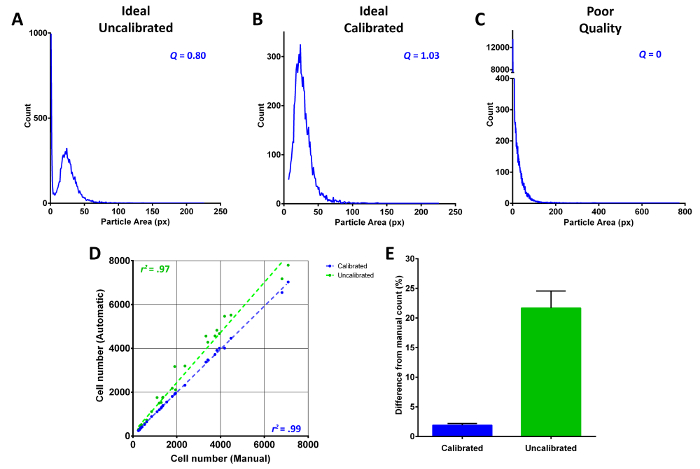
Figure 3: Calibration and validation of migration assay counter. (A) Typical example of an uncalibrated image of high quality. (B) The calibrated version of (A) using migration assay counter's Recount > 'Suggested size'. (C) A typical frequency plot of a low image quality membrane with significant background stain or overall darkness. (D) A scatter plot comparing manual counts using the ImageJ plugin Cell Counter and migration assay counter automated counts. (E) A bar graph showing the average percent difference of calibrated versus uncalibrated automated counts. Data are Mean ± SE. P < 0.0001, n = 30, unpaired t-test with Welch's correction. The same images were used for D and E. Calibration of both was done with the Recount > 'Suggested size' and Recount > 'Manual settings' to further refine the minimum size counted and color threshold. No manual count adjustments were made using the 'Open image with counts' function. Please click here to view a larger version of this figure.
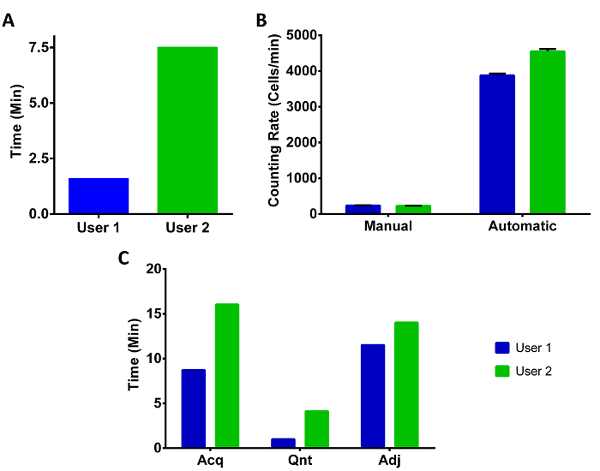
Figure 4: Timing of Cell Concentration Counter and migration assay counter usage. (A) Comparison of CCC calibration times (steps 1 and 2) between User 1 (CCC/TC expert) and User 2 (CCC/TC novice). (B) Manual cell counting times were averaged over five trials and expressed in cells counted per min. Automatic counts were calculated by averaging times to capture nine images of a hemocytometer chamber and counted with CCC. Cell concentration used was ~1.15 x 106 cells/mL. Data are Mean ± SE. n = 5. (C) Transwell Counter timings were compared over three categories: acquisition (Acq; steps 7 and 8), quantification (Qnt; steps 10.1-10.3.3 using default configuration settings), and manual adjustment (Adj; steps 10.4-1.4.2). The membranes quantified had a high degree of background staining and debris in order to necessitate substantial manual adjustment for accurate counts. Please click here to view a larger version of this figure.
