Rescue and Characterization of Recombinant Virus from a New World Zika Virus Infectious Clone
Summary
This protocol describes the recovery of infectious Zika virus from a two-plasmid infectious cDNA clone.
Abstract
Infectious cDNA clones allow for genetic manipulation of a virus, thus facilitating work on vaccines, pathogenesis, replication, transmission and viral evolution. Here we describe the construction of an infectious clone for Zika virus (ZIKV), which is currently causing an explosive outbreak in the Americas. To prevent toxicity to bacteria that is commonly observed with flavivirus-derived plasmids, we generated a two-plasmid system which separates the genome at the NS1 gene and is more stable than full-length constructs that could not be successfully recovered without mutations. After digestion and ligation to join the two fragments, full-length viral RNA can be generated by in vitro transcription with T7 RNA polymerase. Following electroporation of transcribed RNA into cells, virus was recovered that exhibited similar in vitro growth kinetics and in vivo virulence and infection phenotypes in mice and mosquitoes, respectively.
Introduction
Zika virus (ZIKV; Family Flaviviridae: Genus Flavivirus) is a mosquito-borne flavivirus that arrived in Brazil in 2013-14 and was subsequently associated with a massive outbreak of febrile illness that spread throughout the Americas1. In addition, ZIKV has been linked with severe disease outcomes, such as Guillain-Barré syndrome in adults and microcephaly in fetuses and neonates2. Little was known about ZIKV before its rapid spread in the western hemisphere. This included a lack of molecular tools, thus hindering mechanistic research. Molecular tools for viruses, such as infectious cDNA clones, facilitate vaccine and antiviral therapeutic development, and allow for the assessment of viral genetic factors related to differential viral pathogenesis, immune response and viral evolution.
Flavivirus infectious clones are known to be highly unstable in bacteria due to cryptic prokaryotic promoters present in their genomes3. Several approaches have been used to ameliorate this problem; including insertion of tandem repeats upstream of viral sequences4, mutation of putative prokaryotic promoter sequences5, splitting the genome into multiple plasmids6, low-copy number vectors (including bacterial artificial chromosomes)7,8 and insertion of introns in the viral genome9. One full-length system without modifications has been described for ZIKV; however, this clone appeared to be attenuated in cell culture and in mice10. Other groups have engineered introns into the ZIKV genome, allowing for disruption of unstable sequences in bacteria that can be spliced out in mammalian cells in vitro for the production of infectious virus11,12. Additionally, a PCR-based system entitled Infectious-Subgenomic-Amplicons has been used successfully to rescue the prototype MR766 strain of ZIKV13. The approach described here requires no foreign sequences, but rather disrupts the genome at the region of high instability by using multiple plasmids, which has previously been used successfully with yellow fever6, dengue14,15 and West Nile viruses16. Furthermore, the addition of the hepatitis D ribozyme sequence at the viral genomic termini facilitates the creation of an authentic 3' end without the need for the addition of a linearization site. Additionally, both plasmids are constructed in a low-copy number vector (pACYC177, ~15 copies per cell) to mitigate any residual toxicity17. The virus recovered displays a growth profile comparable to parental virus in in vitro growth curves in 8 cell lines comprising a variety of cell-types derived from both mammals and insects, and has exhibited identical pathogenic profiles in mice and infection, dissemination and transmission rates in mosquitoes18.
Herein, we detail a protocol describing how to grow the infectious clone plasmids, generate full-length viral RNA (the viral genome) in vitro, and recover infectious virus in cell culture. First, we describe the propagation of plasmids in bacteria or bacteria-free amplification using rolling circle amplification (RCA). Next, we show how the two plasmids are digested and then subsequently ligated together to generate full-length virus. Finally, we describe the production of transcribed RNA and its subsequent electroporation into Vero cells, followed by titration of recovered virus (Figure 1). The approach described is rapid, allowing for recovery of infectious virus stocks in 1-2 weeks.
Protocol
1. Transform and Recovery of Infectious Clone Plasmids
- Transform both plasmids (separately) using a commercial transformation protocol (e.g., NEB 5 Minute Transformation Protocol) with some modifications. Both plasmids contain a gene encoding for ampicillin resistance, therefore use ampicillin or carbenicillin for selection. Carbenicillin is preferred, as it is more stable.
- Remove cells (See Materials Table) from -80 °C freezer and thaw on ice for 5-10 min. Prewarm lysogeny broth (LB) (containing 10 g/L NaCl and 25 µg/mL carbenicillin; called LB-carb) plates and the broth with catabolite repression (SOC (without antibiotics) – coming with the cells) at 37 °C.
- Add between 100 pg-10 ng in 1 µL of plasmid DNA to a tube containing 50 µL of competent cells; mix gently by flicking the tube.
- Incubate tubes on ice for 5 min.
- Heat shock tubes at 42 °C for 30 s in a water bath. Return to ice for 2 min immediately following heat shock.
- Pipette 950 µL of room temperature SOC into each tube and mix by pipetting. Spread 100 µL of the bacterial mixture onto a LB-carb plate and incubate overnight at 37 °C (roughly 12-14 h).
- Remove plates from 37 °C incubator after 12-14 h incubation. Place plates in a dark drawer at room temperature to allow colonies to continue to grow (roughly 8-9 h).
- Using 5 mL of Terrific broth (TB, containing 25 µg/mL carbenicillin, called TB-carb), grow 5-10 small colonies for each plasmid overnight at 30 °C in a bacterial shaker (roughly 14-16 h).
NOTE: Small colonies are an indicator that the plasmid is intact; colonies with plasmids containing deletions, rearrangements or mutations (herein called instability events) will grow faster, and thus be bigger. Therefore, one should attempt to pick the smallest colonies on the plate, specifically not selecting the largest colonies present. Some medium size colonies may also be selected for completeness. The lower temperature used reduces the probability that the cells will overgrow (OD600 of greater than 0.6-0.8). As most researchers perform overnight growth, it allows for more control and reduced chance of overgrowth. - Check liquid cultures for turbidity (roughly OD600 of 0.6-0.8). Place any turbid tubes at 4 °C and allow other tubes to become turbid by growing for longer.
NOTE: Do not grow bacteria to OD600 values greater than recommended, as plasmid instability events may occur. - Extract plasmid DNA from bacteria with a commercial plasmid miniprep kit (See Materials Table). Elute in 15 µL of pre-heated (to 70 °C in a heating block) elution buffer. Allow the elution buffer to incubate on the column for 5 min before centrifugation.
NOTE: Preserve at least 1 mL of this starter culture at 4 °C for further use in the next step. - Digest 10 µL of the recovered plasmids to assess whether plasmid instability events occurred during culture.
NOTE: Digest p1 with SalI/NheI and p2 with BamHI/HindIII at 37 °C for 1 h. Confirm the presence of the correct bands following digestion (Figure 2a) by gel electrophoresis and proceed to step 2. If preparing plasmid DNA by rolling circle amplification (RCA), reserve some of the miniprep eluate to serve as a template.
2. Preparation of Sufficient Plasmid DNA for Ligation
NOTE: Ligation and subsequent electroporation requires a sufficient amount of plasmid DNA that must be generated via either maxiprep or RCA. While maxiprep is the traditionally used approach, RCA offers the advantage of not requiring bacteria, thus removing the potential for plasmid-induced toxicity in bacteria that can result in instability events.
- Perform the following maxiprep procedure.
- For each plasmid that demonstrated the correct restriction enzyme digestion pattern in the previous section, add 500 µL of starter culture to 1 L of TB-Carb (25 µg/mL carbenicillin) broth and shake overnight at 30 °C (roughly 14-16 h, roughly OD600 of 0.6-0.8).
NOTE: Do not allow the culture to grow over this OD600 value. This will potentially result in instability events within the plasmids. - Harvest the bacteria and perform maxipreps per manufacturer's protocol (See Materials Table).
- For each plasmid that demonstrated the correct restriction enzyme digestion pattern in the previous section, add 500 µL of starter culture to 1 L of TB-Carb (25 µg/mL carbenicillin) broth and shake overnight at 30 °C (roughly 14-16 h, roughly OD600 of 0.6-0.8).
- Using the saved miniprep from step 1.6, perform the following for the RCA procedure.
- Transfer 1 µL of plasmid DNA to a tube containing 50 µL of sample buffer.
- Heat-denature the sample at 95 °C for 3 min, and then incubate at 4 °C until ready to use.
- Prepare the commercial amplification premix (See Materials Table). Combine 50 µL of reaction buffer with 2 µL of enzyme mix in a tube set on ice.
NOTE: The amplification premix should be kept on ice until ready for use. It is convenient to prepare a master mix by combining sufficient reagents for the required number of amplification reactions. Any unused premix must be discarded. - Transfer 50 µL of prepared amplification premix to the denatured sample.
- Incubate the reaction tubes at 30 °C for 18 h.
- Incubate the tubes at 65 °C for 10 min to inactivate the ø29 DNA polymerase.
- Store the reaction tubes at 4 °C or -20 °C until ready for use.
NOTE: At this point, plasmid DNA prepared via both methods should be quantified (using absorbance at 260 nm) and the ZIKV genome should be Sanger sequenced entirely to confirm no instability events (deletions and rearrangements) occurred during preparation.
| Primer Name | Sequence (5' – 3') |
| ZIKV PRVABC59 1For. | AGTTGTTGATCTGTGTGAATCAGAC |
| ZIKV Conserved 632For. | GCCCTATGCTGGATGAGG |
| ZIKV Conserved 692Rev. | GGTTCCGTACACAACCCAAGTTG |
| ZIKV Conserved 1313Rev. | CTCCCTTTGCCAAAAAGTCCACA |
| ZIKV Conserved 1201For. | CCAACACAAGGTGAAGCCTAC |
| ZIKV Conserved 1663For. | GCAGACACCGGAACTCCACACT |
| ZIKV Conserved 2008For. | CAGATGGCGGTGGACATGC |
| ZIKV Conserved 2350Rev. | GTGAGAACCAGGACATTCCTCC |
| ZIKV Conserved 2605For. | TACAAGTACCATCCTGACTCCC |
| ZIKV Conserved 3499Rev. | GCCTTATCTCCATTCCATACCA |
| ZIKV Conserved 3961Rev. | TTGCCAACCAGGCCAAAG |
| ZIKV Conserved 4132For. | CATTTGTCATGGCCCTGG |
| ZIKV Conserved 4561Rev. | CTATTGGGTTCATGCCACAGAT |
| ZIKV Conserved 4665For. | GACCACAGATGGAGTGTACAGAGT |
| ZIKV Conserved 5189Rev. | AAGACAGTTAGCTGCTTCTTCTTCAG |
| ZIKV Conserved 5219For. | GAGAGTTCTTCCTGAAATAGTCCGTGA |
| ZIKV Conserved 6086Rev. | CTTGCTTCAAGCCAGTGTGC |
| ZIKV Conserved 6119For. | GCCTCATAGCCTCGCTCTATCG |
| ZIKV Conserved 6721Rev. | CCATTCCAAAGCCCATCTTCCC |
| ZIKV Conserved 6769For. | CCAGCCAGAATTGCATGTGTCC |
| ZIKV Conserved 7209Rev. | ATCATTAGCAGCGGGACTCCAA |
| ZIKV Conserved 7343For. | GTTGTGGATGGAATAGTGGT |
| ZIKV Conserved 8243For. | TGCCCATACACCAGCACTATGA |
| ZIKV PRVABC59 8893Rev. | TGCATTGCTACGAACCTTGTTG |
| ZIKV conserved 9133For. | AGCCCTTGGATTCTTGAACGAGGA |
| ZIKV PRVABC59 9673Rev. | AACGCAATCATCTCCACTGACT |
| ZIKV Conserved 9686For. | GATGATAGGTTTGCACATGCC |
| ZIKV Conserved 10321Rev. | GTCCATGTACTTTTCTTCATCACCTAT |
| ZIKV Conserved 10455For. | CAGGAGAAGCTGGGAAACC |
| ZIKV Conserved 10621Rev. | CAGATTGAAGGGTGGGGAAGGTC |
Table 1: Primers for sequencing cDNA clones. Plasmids can be sequenced entirely with the primers listed. Primers marked as "conserved" indicate that they are likely able to sequence/amplify all genotypes of ZIKV. Primers labelled as "PRVABC59" are specific to this strain but may also work for other Asian genotype strains and possibly other genotypes.
3. Preparation of In Vitro Transcribed RNA
- Set up digestion reaction of either maxiprep or RCA prepared DNA.
- For p1 digestion, mix 10 µL of 10x reaction buffer, 3 µL of ApaLI, 3 µL of BamHI-HF, 3 µL of rSAP or CIP, and 3.3 µg of p1 DNA. Add ddH2O to 100 µL.
- For p2 digestion, mix 10 µL of 10x reaction buffer, 3 µL of ApaLI, 3 µL of EcoRI-HF, and 13.3 µg of p2 DNA. Add ddH2O to 100 µL.
- Incubate at 37 °C for 1-2 h.
- Purify using a commercial gel and PCR clean-up kit (See Materials Table). Elute in 30 µL of elution buffer pre-heated to 70 °C in a heating block (incubate the column for 5 min before spinning).
NOTE: Keep 1 µL to run on a gel later.
- Prepare ligation reaction.
- Combine 10 µL of 10x T4 DNA ligation reaction buffer, 3 µL of T4 DNA ligase (400 U/µL), 29 µL of purified p1, and 29 µL purified p2. Add ddH2O to 100 µL.
- Incubate at room temperature for 2 h or 16 °C overnight.
- Purify using a commercial gel and PCR clean-up kit (See Materials Table). Elute in 10 µL of elution buffer pre-heated to 70 °C in a heating block (incubate column for 5 minutes before spinning).
NOTE: Keep 1 µL to run on a gel later.
- Run undigested plasmids, digested plasmids, and ligation on an agarose gel. The ligation product should have a higher molecular weight band (around 11 kb) than either digested plasmid alone, indicating the ligation was successful (Figure 2b).
- Set up T7 in vitro transcription reaction.
- Combine 10 µL 2x ARCA/NTP Mix, 2 µL of T7 RNA polymerase mix, and 8 µL of purified ligation reaction for a final volume of 20 µL.
NOTE: The ARCA included in the mix is a cap analog that is exclusively incorporated in the correct orientation. Standard cap analogs can result in only ~50% of the transcripts having a cap in the correct orientation. - Incubate for 4 h at 37 °C.
- Measure RNA concentration via a UV spectrophotometer. Optionally, run a denaturing agarose gel to confirm the length of the RNA transcript.
- Combine 10 µL 2x ARCA/NTP Mix, 2 µL of T7 RNA polymerase mix, and 8 µL of purified ligation reaction for a final volume of 20 µL.
4. Rescue of Infectious Virus by Electroporation
- days prior to electroporation, prepare 1 T182 flask ofVero cells (between T150 and T182 is acceptable; grown in DMEM + 10% Fetal bovine serum (FBS)) per construct to be recovered. Include 1 T182 as a mock transfection control. Cells should be used at 70-80% confluency.
- When cells are ready, place 12 mL/reaction of DMEM + 15% FBS + 10 mM HEPES in a water bath heated to 37°C.
- Detach cells by trypsinization with a standard protocol, and recover cells in media with 10% FBS to inactivate trypsin; centrifuge cells at 150 x g for 5 min at 4 °C.
- Wash cells in 1x phosphate-buffered saline (PBS), pelleting cells at 150 x g for 5 min at 4 °C. Repeat this step twice for a total of three washes.
- Resuspend cells in 200 µL of RPMI 1640 + 10 mM HEPES (sterile) per # of samples. Transfer 200 µL of cell suspension to a sterile tube. Cells should be ~0.5 x 107 cells/mL.
- Add 20 µL of water (mock neg. control) or 20 µL of RNA (preferably 5-10 µg) produced in step 3.4 to each set of cells. Mix gently by flicking the tube and transfer the contents of the tube to a 2 mm gap electroporation cuvette.
- Set an electroporator to 170 V LV, omega (resistance) at 0 and 950 µF (roughly to 625 V/cm and 20 ms time constant for other machines). Set resistance to the lowest available setting, either 0 or ∞ if available.
- Pulse once and immediately add 600 µL of DMEM +15 %FBS +10 mM HEPES that was warmed in step 4.2. Rinse inside of cuvette thoroughly to remove all cells. Add cells to a T75 tissue culture flask containing 10 mL of pre-warmed media. Remove additional media from cuvette by using the dropper that is included with the cuvette. Be careful to maintain sterility.
- Swirl flask to mix media and cells and place in 37 °C incubator.
- Check the next day for cell viability, and do so every day until 50-75% cell death is observed. Cell death is observed by comparing to the mock electroporated control.
NOTE: ZIKV cytopathic effect (CPE) is quite pronounced in Vero cells, resulting in rounded up cells that detach and float in the culture medium. We have observed a range of 8-10 days as being ideal for harvest. - Harvest supernatant in a conical tube and centrifuge to remove cellular debris at >3,000 x g for 10 min at 4 °C.
- Remove clarified supernatant to a new tube and supplement with a final concentration of 20% FBS (from 100% stock). Additionally, add sterile HEPES at a final concentration of 10 mM as a buffering agent to preserve virus infectivity while frozen (from a 1 M sterile stock).
- Mix virus by vortexing and aliquot into screw cap vials. Store at -80 °C for future use.
5. Titration of Recovered Virus
- Seed the appropriate number of 6- or 12-well plates of Vero cells the day before titration.
- Remove virus from the freezer and make serial 10-fold dilutions in DMEM+ 2%FBS in tubes or 96-well plates.
NOTE: The expected titer from recovered clones is between 1 x 106 and 5 x 107 PFU/mL. - Add 200 or 400 µL of each dilution in duplicate to a well of a 12- or 6-well plate, respectively.
- Place plates in 37 °C incubator and rock every 15 min, for a total of ~1-1.5 h adsorption period.
- Remove viral inoculum and add 1 mL of (12-well) or 2 mL (6-well) DMEM-Tragacanth overlay to each well.
NOTE: It is possible to use agarose, agar, methylcellulose or other overlay media here. - Incubate cells in 37 °C incubator for 5 days. The time for proper plaque formation will vary depending on the overlay used.
- To fix cells, remove plates from incubator and decant overlay gently into disinfectant.
- Add sufficient 20% ethanol/0.1% crystal violet (CV) to each well to cover and swirl to mix.
NOTE: Many other fixatives exist and can be used here. Additionally, if using an agarose or agar overlay, a second overlay can be used with in conjunction with neutral red to visualize plaques without fixation. 20% ethanol has shown to be effective in inactivating enveloped viruses in previous studies; do not use this amount for non-enveloped viruses as they will not be inactivated19. - Incubate plates for 30-60 min at room temperature (in a class II biosafety cabinet); discard CV (per local regulations) and wash plates with tap water.
- Allow plates to dry, and manually count plaques.
NOTE: Reference 20 can be used as an additional reference for performing plaque assays.
Representative Results
The protocol described here allows for the recovery of infectious clone-derived Zika virus. Manipulating the two-plasmid infectious clone system is straightforward when performed with care, as compared to full-length versions which are highly unstable (data not shown). After digestion and ligation of the two distinct pieces, capped RNA is produced using in vitro transcription with T7 polymerase which is then electroporated into Vero cells (Figure 1). Correct plasmid sequence can be monitored using restriction digestion as a proxy following miniprep (Figure 2a) and via Sanger sequencing after large-scale DNA production with RCA or maxiprep. After confirmation of the clones by sequencing, recovery of infectious virus requires only digestion, ligation, in vitro transcription and finally electroporation. These steps can be performed in one day. Correct digestion and ligation can be monitored by agarose gel electrophoresis (Figure 2b), allowing for troubleshooting, if necessary.
As shown in Figure 3, infectious clone-derived virus replicates to similar levels as the parental isolate in vitro in several cell lines, suggesting that the recovered virus from the cDNA replicates similarly to primary isolate. Additionally, no significant differences were observed between the infectious clone-derived virus and the parental isolate in rates of survival in mice or rates of infection, dissemination and transmission in Aedes aegypti mosquitoes (Figure 4).
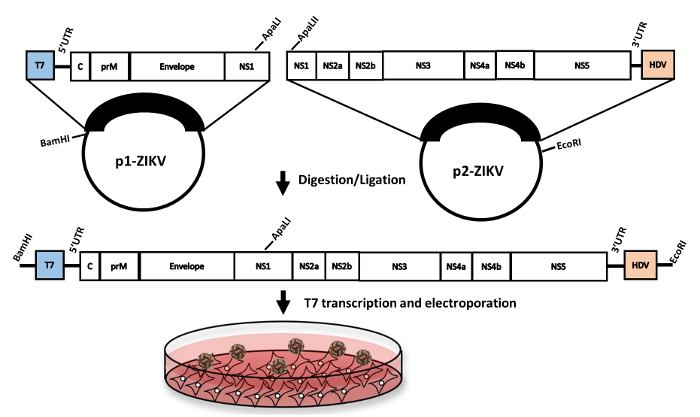
Figure 1: Workflow of ZIKV rescue from a two-plasmid cDNA clone. The genome of ZIKV strain PRVABC59 was cloned in two-separate pieces into a pACYC177 plasmid backbone. The two plasmids were then digested and ligated with T4 DNA ligase. Capped-infectious RNA was then produced via in vitro transcription followed by electroporation into Vero cells. (Figure adapted from reference21). Please click here to view a larger version of this figure.
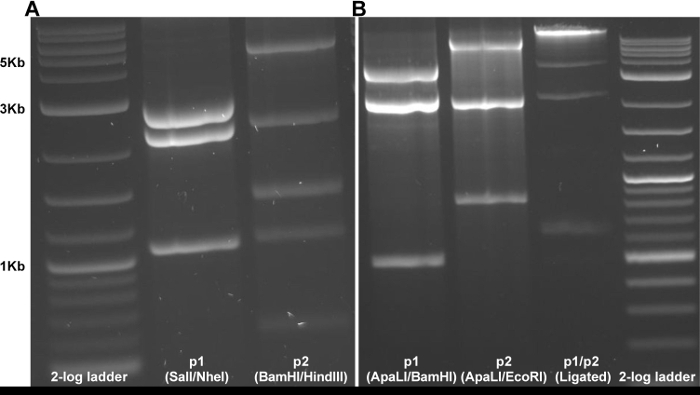
Figure 2: Gel electrophoresis to monitor digestion and ligation of cDNA clone. (A) Initial restriction digestion of miniprep derived plasmids to assess genetic stability. (B) Examples of digestion and ligation products during recovery of infectious virus from the two-plasmid clone system. Please click here to view a larger version of this figure.
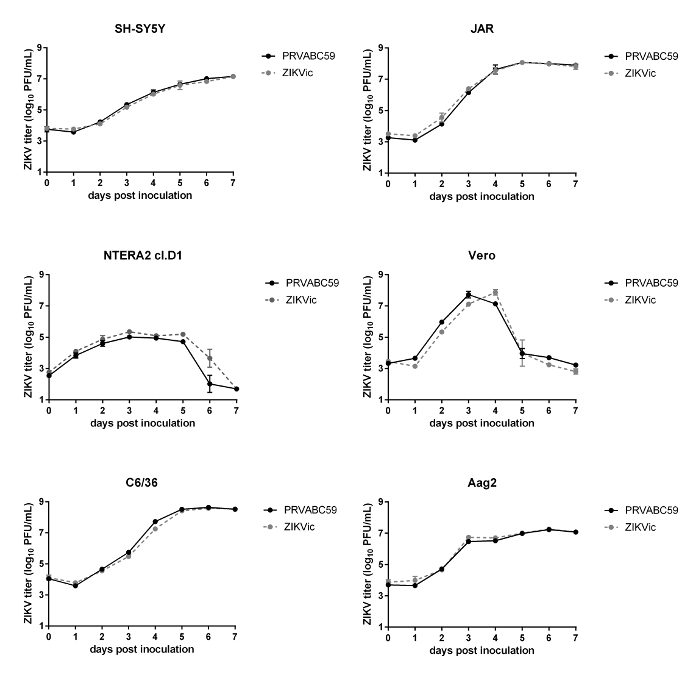
Figure 3: In vitro growth kinetics of clone-derived virus. Growth kinetics of wild type PRVABC59 and infectious clone derived virus on human (SH-SY5Y, Jar and NTERA2 cI.D1), mosquito (C6/36, Aag2) and non-human primate (Vero) cell lines were assessed by plaque assay. n = 3 Error bars represent standard deviation from the mean. (Figure adapted from reference21). Please click here to view a larger version of this figure.
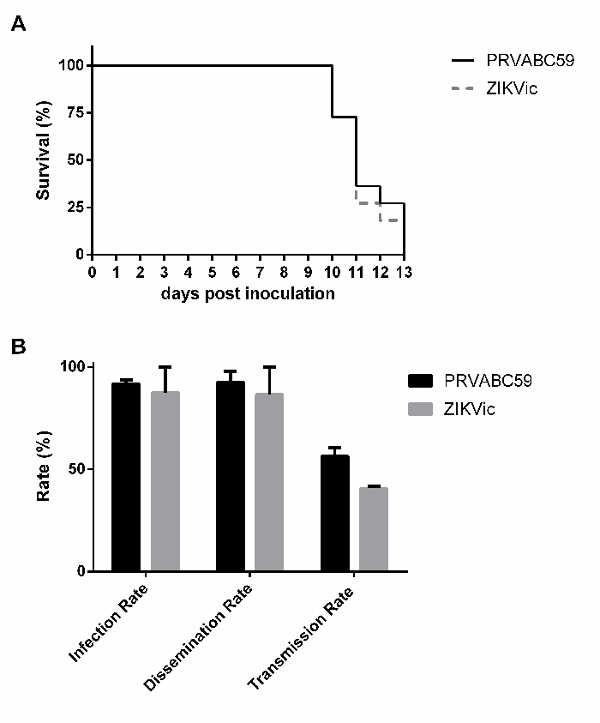
Figure 4: In vivo characterization of clone-derived virus in mice and mosquitoes. (A) 4-week-old male and female interferon alpha, beta and gamma receptor knockout,AG129, mice were intraperitoneally inoculated with 1,000 PFU of PRVABC59 or the infectious clone-derived virus and monitored over time for survival (n=11). Aedes aegypti mosquitoes were given an infectious bloodmeal containing ZIKV and dissected 14 days later. (B) Plaque assays were used to assess mosquito bodies (infection), legs (dissemination) or salivary secretions (transmission) for infectious virus. Rates are presented as the percent of the total mosquitoes that were tested (Figure adapted from reference21). Please click here to view a larger version of this figure.
Discussion
Here we describe a method for the recovery of a bipartite infectious cDNA clone system for ZIKV. Previously described clones for ZIKV suffer from either attenuation or require the addition of introns, making plasmids larger and preventing rescue in insect cells. Infectious virus can be recovered using the two-plasmid clone system in either mammalian or insect cells (data not shown). In addition, virus recovered from this system behaves similarly to wild-type virus in several cell lines, in an immunocompromised mouse model and in mosquitoes.
Due to the presence of cryptic bacterial promoters in flavivirus genomes3, full-length flavivirus clones are difficult to work with, commonly resulting in mutations, insertions, deletions or rearrangements in plasmids. We sought to alleviate these concerns by separating the ZIKV genome into two plasmids, disrupting the toxic sequences that are present in the viral NS1 gene. This resulted in a more stable system that is also amenable to incorporating changes. Using this clone system, we have introduced several point mutations in the ZIKV genome and inserted foreign sequences in the 3′ UTR (data not shown). This system also provides an ideal starting point for studying genetic determinants of pathogenesis, construction of vaccine candidates and development of reporter viruses. Additionally, the clone-derived virus has proven to be an important tool for studying virus evolution22.
The methods described here can also be applied to new two-plasmid clone systems for other ZIKV strains or other flaviviruses, with some modifications. An important consideration for creating new virus clone systems using this approach is identifying a naturally occurring restriction enzyme site in the viral genome near the envelope-NS1 gene junction. If a suitable site cannot be found, then it is possible to generate one artificially via PCRmutagenesis. Alternatively, we have successfully rescued infectious virus by using Gibson assembly, if overlapping segments can be generated via restriction digestion (data not shown). Furthermore, it is necessary to include either a restriction enzyme cleavage site or a ribozyme sequence immediately downstream of the viral genome to ensure an authentic 3’UTR.
By using a two-plasmid approach, we found that a stable and relatively straightforward cDNA clone system for ZIKV could be developed. The infectious virus recovered from the clone possessed wild-type characteristics in vitro and in vivo. Additionally, this approach can be adapted for any flavivirus, facilitating reverse genetics approaches for other relevant viruses.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors would like to thank Kristen Bullard-Feibelman, Milena Veselinovic and Claudia Rückert for their assistance in characterizing the clone-derived virus. This work was supported in part by grants from the National Institute of Allergy and Infectious Diseases, NIH under grants AI114675 (BJG) and AI067380 (GDE).
Materials
| NEB Stable CompetentE. coli | New England BioLabs | C3040H | |
| Carbenicillin, Disodium Salt | various | ||
| Zyppy Plasmid Miniprep Kit | Zymo Research | D4036 | |
| ZymoPURE Plasmid Maxiprep Kit | Zymo Research | D4202 | |
| SalI-HF | New England BioLabs | R3138S | 20,000 units/ml |
| NheI-HF | New England BioLabs | R3131S | 20,000 units/ml |
| ApaLI | New England BioLabs | R0507S | 10,000 units/ml |
| EcoRI-HF | New England BioLabs | R3101S | 20,000 units/ml |
| BamHI-HF | New England BioLabs | R3136S | 20,000 units/ml |
| HindIII-HF | New England BioLabs | R3104S | 20,000 units/ml |
| illustra TempliPhi 100 Amplification Kit | GE Healthcare Life Sciences | 25640010 | |
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | 740609.5 | |
| Shrimp Alkaline Phosphatase (rSAP) | New England BioLabs | M0371S | 1,000 units/ml |
| Alkaline Phosphatase, Calf Intestinal (CIP) | New England BioLabs | M0290S | 10,000 units/ml |
| T4 DNA Ligase | New England BioLabs | M0202S | 400,000units/mL |
| HiScribe T7 ARCA mRNA Kit | New England BioLabs | E2065S | |
| Vero cells | ATCC | CCL-81 | |
| ECM 630 High Throughput Electroporation System | BTX | 45-0423 | Other machines are acceptable. |
| LB Broth with agar (Miller) | Sigma | L3147 | Can be homemade as well. |
| Terrific Broth | Sigma | T0918 | Can be homemade as well. |
| Petri Dish | Celltreat | 229693 | |
| Culture Tubes | VWR International | 60818-576 | |
| T75 flasks | Celltreat | 229340 | |
| T182 flasks | Celltreat | 229350 | |
| 1x PBS | Corning | 21-040-CV | |
| RPMI 1640 with L-glutamine | Corning | 10-040-CV | |
| DMEM with L-glutamine and 4.5 g/L glucose | Corning | 10-017-CV | |
| Fetal Bovine Serum (FBS) | Atlas Biologicals | FP-0500-A | |
| Tragacanth Powder | MP Bio | MP 104792 | |
| Crystal Violet | Amresco | 0528-1006 | |
| Ethanol Denatured | VWR International | BDH1156-1LP | |
| 6 well plate | Celltreat | 229106 | |
| 12 well plate | Celltreat | 229111 | |
| Sequencing Oligos | IDT | see table 1 | |
| Qubit 3.0 | ThermoFisher | Qubit 3.0 | other methods are acceptable. |
| Qubit dsDNA BR Assay Kit | ThermoFisher | Q32850 | other methods are acceptable. |
| Qubit RNA HS Assay Kit | ThermoFisher | Q32852 | other methods are acceptable. |
| Class II Biosafety Cabinet | Varies | N/A | This is necessary for live-virus work. |
References
- Kindhauser, M. K., Allen, T., Frank, V., Santhana, R. S., Dye, C. Zika: the origin and spread of a mosquito-borne virus. Bull World Health Organ. 94 (9), 675C-686C (2016).
- Oehler, E., et al. Zika virus infection complicated by Guillain-Barre syndrome–case report, French Polynesia, December 2013. Euro Surveill. 19 (9), (2014).
- Li, D., Aaskov, J., Lott, W. B. Identification of a cryptic prokaryotic promoter within the cDNA encoding the 5′ end of dengue virus RNA genome. PLoS One. 6 (3), e18197 (2011).
- Pu, S. Y., et al. A novel approach to propagate flavivirus infectious cDNA clones in bacteria by introducing tandem repeat sequences upstream of virus genome. J Gen Virol. 95 (Pt 7), 1493-1503 (2014).
- Pu, S. Y., et al. Successful propagation of flavivirus infectious cDNAs by a novel method to reduce the cryptic bacterial promoter activity of virus genomes. J Virol. 85 (6), 2927-2941 (2011).
- Rice, C. M., Grakoui, A., Galler, R., Chambers, T. J. Transcription of infectious yellow fever RNA from full-length cDNA templates produced by in vitro ligation. New Biol. 1 (3), 285-296 (1989).
- Yun, S. I., Kim, S. Y., Rice, C. M., Lee, Y. M. Development and application of a reverse genetics system for Japanese encephalitis virus. J Virol. 77 (11), 6450-6465 (2003).
- Gualano, R. C., Pryor, M. J., Cauchi, M. R., Wright, P. J., Davidson, A. D. Identification of a major determinant of mouse neurovirulence of dengue virus type 2 using stably cloned genomic-length cDNA. J Gen Virol. 79 (Pt 3), 437-446 (1998).
- Johansen, I. E. Intron insertion facilitates amplification of cloned virus cDNA in Escherichia coli while biological activity is reestablished after transcription in vivo. Proc Natl Acad Sci U S A. 93 (22), 12400-12405 (1996).
- Shan, C., et al. An Infectious cDNA Clone of Zika Virus to Study Viral Virulence, Mosquito Transmission, and Antiviral Inhibitors. Cell Host Microbe. 19 (6), 891-900 (2016).
- Schwarz, M. C., et al. Rescue of the 1947 Zika Virus Prototype Strain with a Cytomegalovirus Promoter-Driven cDNA Clone. mSphere. 1 (5), (2016).
- Tsetsarkin, K. A., et al. A Full-Length Infectious cDNA Clone of Zika Virus from the 2015 Epidemic in Brazil as a Genetic Platform for Studies of Virus-Host Interactions and Vaccine Development. MBio. 7 (4), (2016).
- Gadea, G., et al. A robust method for the rapid generation of recombinant Zika virus expressing the GFP reporter gene. Virology. 497, 157-162 (2016).
- Kapoor, M., Zhang, L., Mohan, P. M., Padmanabhan, R. Synthesis and characterization of an infectious dengue virus type-2 RNA genome (New Guinea C strain). Gene. 162 (2), 175-180 (1995).
- Messer, W. B., et al. Development and characterization of a reverse genetic system for studying dengue virus serotype 3 strain variation and neutralization. PLoS Negl Trop Dis. 6 (2), e1486 (2012).
- Kinney, R. M., et al. Avian virulence and thermostable replication of the North American strain of West Nile virus. J Gen Virol. 87 (Pt 12), 3611-3622 (2006).
- Chang, A. C., Cohen, S. N. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol. 134 (3), 1141-1156 (1978).
- Weger-Lucarelli, J., et al. Development and Characterization of Recombinant Virus Generated from a New World Zika Virus Infectious Clone. J Virol. 91 (1), (2017).
- Roberts, P. L., Lloyd, D. Virus inactivation by protein denaturants used in affinity chromatography. Biologicals. 35 (4), 343-347 (2007).
- Baer, A., Kehn-Hall, K. Viral concentration determination through plaque assays: using traditional and novel overlay systems. J Vis Exp. (93), e52065 (2014).
- Weger-Lucarelli, J., et al. Development and Characterization of Recombinant Virus Generated from a New World Zika Virus Infectious Clone. J Virol. , (2016).
- Grubaugh, N. D., et al. Genetic Drift during Systemic Arbovirus Infection of Mosquito Vectors Leads to Decreased Relative Fitness during Host Switching. Cell Host Microbe. 19 (4), 481-492 (2016).
