Detailed pipelines for bioinformatic analysis of ribosome profiling data have been described previously 8,9. In addition, several research groups have developed bioinformatics tools for differential gene expression analysis and processing of sequencing data, which are specific for ribosome profiling method 10,11,12,13,14,15,16,17,18. The first step in the analysis of Ribo-Seq data is demultiplexing and trimming the 3' adapter sequence AGATCGGAAGAGCACACGTCT using Cutadapt software 19. Then the sequencing reads are aligned against non-coding RNAs, such as rRNA and tRNA, using Bowtie 20 to remove contaminating sequences, and reads that do not align are then mapped to the yeast genome. Read count per gene can then be assessed by HTseq-count software 21, and differentially expressed genes are identified using DESeq2 22.
Figure 8 shows representative results of quantitative analysis of translation in hbs1Δ 23,24 yeast deletion mutant that were obtained using our protocol. First, we assessed the number and proportion of reads that correspond to non-coding RNA (e.g. rRNAs) obtained after sequencing footprint and mRNA libraries generated for wild-type cells and the hbs1Δ mutant. We found that, even without any rRNA depletion steps (discussed below), from 50 to 60% of all sequenced reads in our footprint libraries correspond to ribosome-protected footprint fragments (Figure 8A). In contrast, only 3% of rRNA-derived fragments were observed in mRNA libraries demonstrating that poly(A) mRNA isolation allows effective elimination of rRNA reads. Together, we were able to obtain more than 10 million footprint reads for each of the footprint samples by sequencing 2 replicates. To assess the variability of library generation and reproducibility of the data, we usually analyze at least two independent biological replicates per each experimental condition. Both footprint and mRNA sample libraries show good reproducibility with Pearson correlation coefficient R ~ 0.99 between matched samples (Figure 8B).
In addition to assessing the level of protein translation at the genome-wide level, ribosome profiling allows measuring changes in translation efficiency between experimental conditions 3. For this, an aliquot of the cell lysate (not treated with ribonuclease) is used for poly(A) mRNA isolation and preparation of an RNA-Seq library. Because both footprint library and RNA-Seq library are prepared under the same controlled conditions and can be traced to each individual replicate, Ribo-Seq and RNA-Seq datasets can be directly compared to identify genes that are regulated by the changes in mRNA transcription, translation efficiency, or by a combined effect. To identify genes that are up- or down-regulated specifically at the level of protein translation in the hbs1Δ mutant, we calculated changes in translation efficiency by dividing footprint rpkm values by mRNA rpkm for each of the genes (Figure 8C).
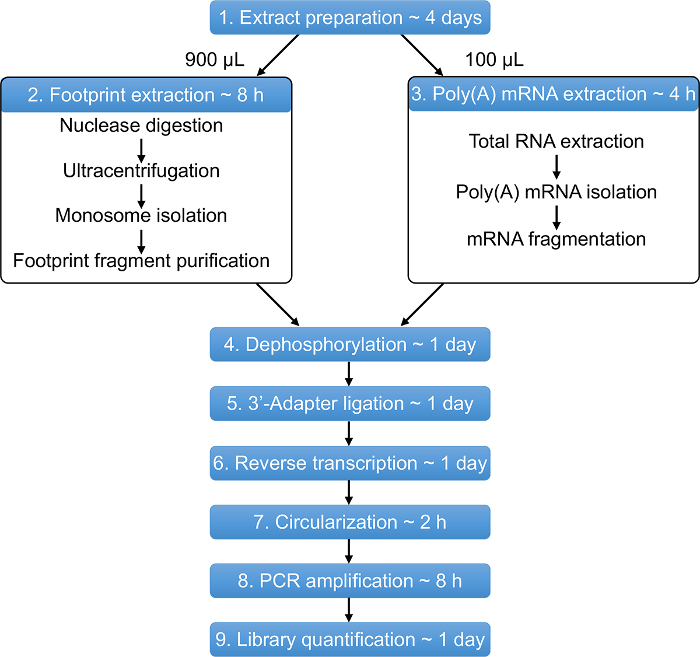
Figure 1: Overview of the Ribo-Seq protocol. The entire protocol can be performed in approximately 11 days. Estimated time for each step is shown. Please click here to view a larger version of this figure.
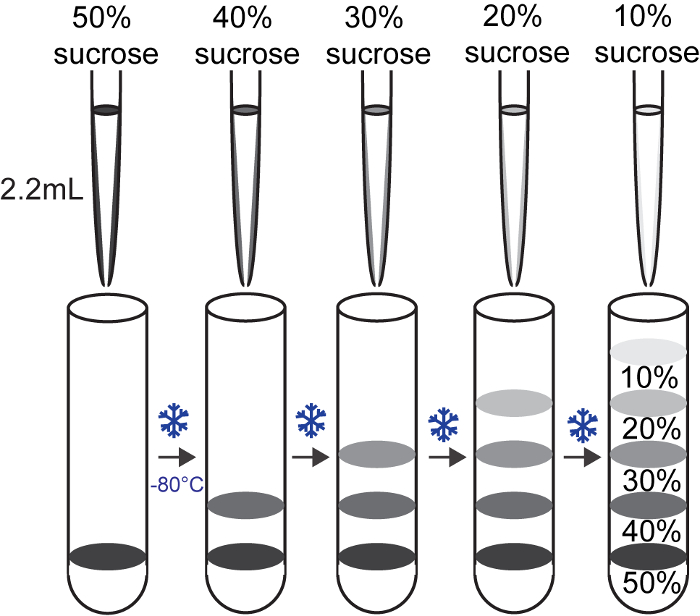
Figure 2: Preparation of sucrose gradients Please click here to view a larger version of this figure.
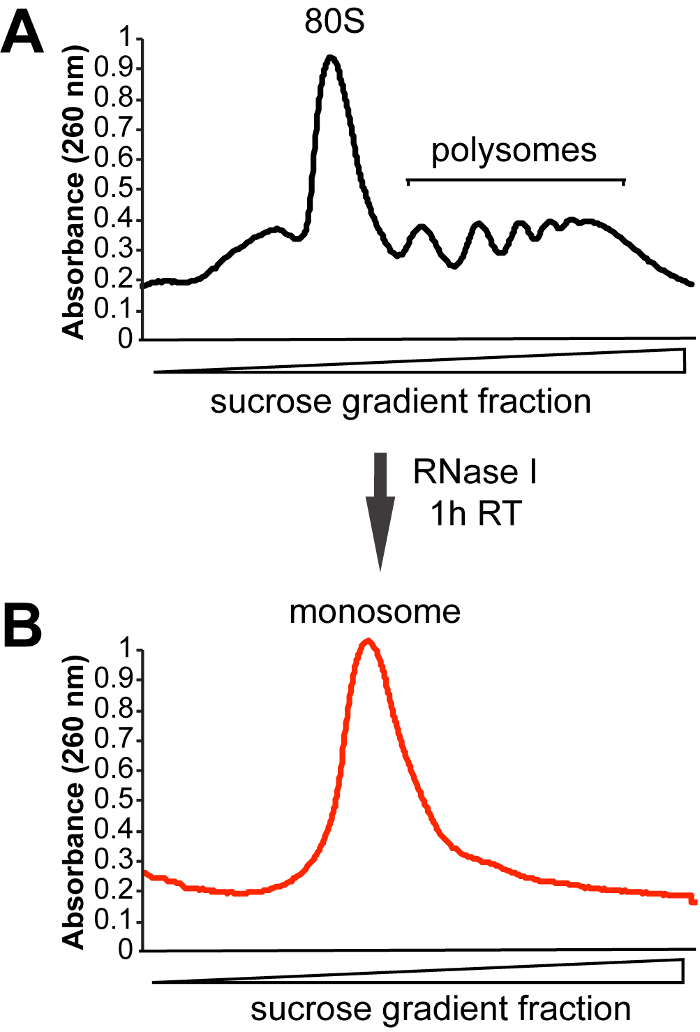
Figure 3: Representative sucrose gradient profiles. (A) Sucrose gradient profile obtained for control (not treated with RNase I) sample. (B) In order to extract ribosome-protected RNA fragments, cell lysates are treated with RNase I for 1 h at room temperature (RT). Fractions corresponding to the monosomal peak are then collected for footprint extraction. Please click here to view a larger version of this figure.
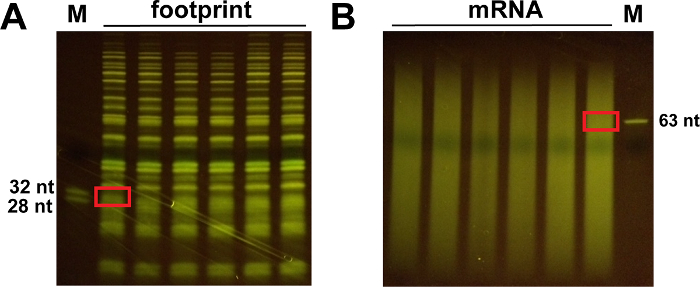
Figure 4: Representative images of the 15% polyacrylamide gels obtained after T4 polynucleotide kinase treatment. (A) The size of the excised gel slice around 28 and 32 nt is shown for footprint samples. (B) Cut the gel slice about 50-70 nt for mRNA samples. Please click here to view a larger version of this figure.
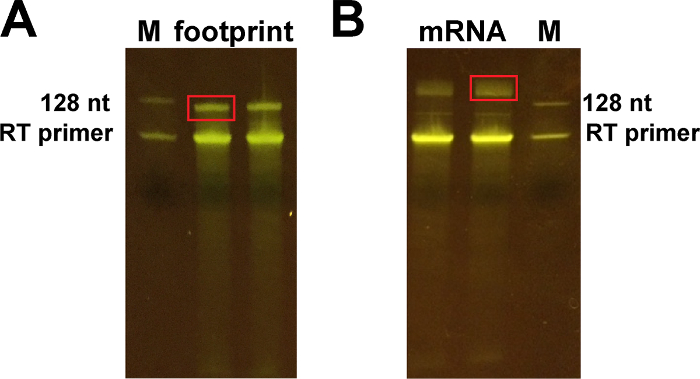
Figure 5: Representative images of the 10% polyacrylamide gels obtained after reverse transcription. (A) Cut the upper band ~ 128 nt for footprint samples, corresponding to the product of reverse transcription. (B) Cut the band around 150-170 nt for the mRNA samples (upper band). The lower bands correspond to the RT primer. Please click here to view a larger version of this figure.
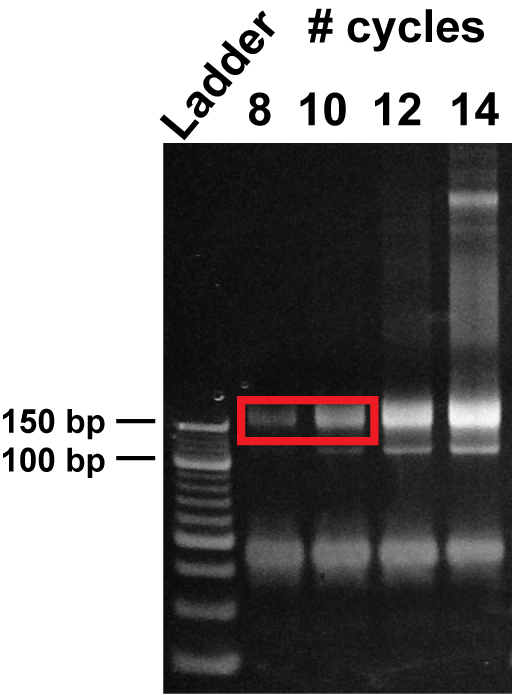
Figure 6: Polyacrylamide gel purification of PCR-amplified libraries. PCR products obtained after 8, 10, 12, and 14 cycles of library amplification were resolved on a non-denaturing 8% TBE gel. The size of the full-length footprint libraries is ~ 150 bp, whereas the size of mRNA libraries is ~170-190 bp. Avoid the lower band that does not contain insert.
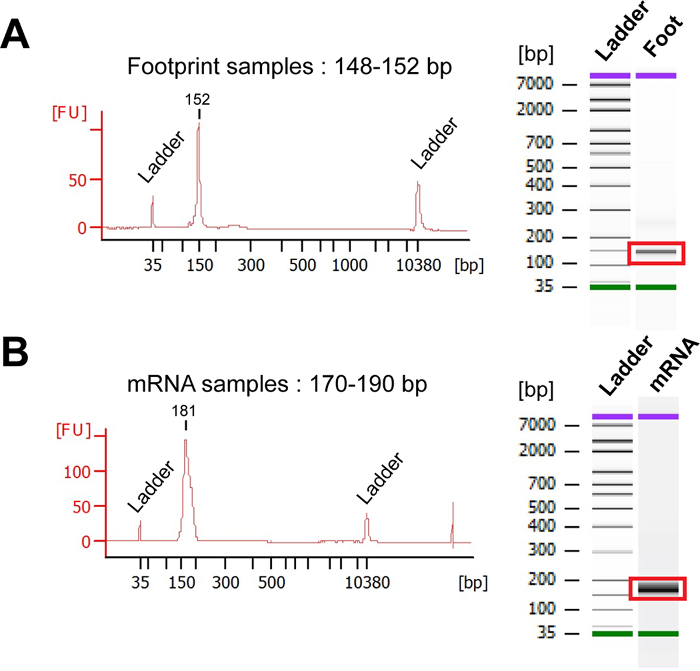
Figure 7: Bioanalyzer analysis. (A) Representative Bioanalyzer profiles of the PCR-amplified footprint library. The average size of the footprint library is expected to be from 148 to 152 nt. (B) Representative Bioanalyzer profiles of the sequencing library obtained for mRNA samples. The expected size of the mRNA library is 170-190 nt. Please click here to view a larger version of this figure.
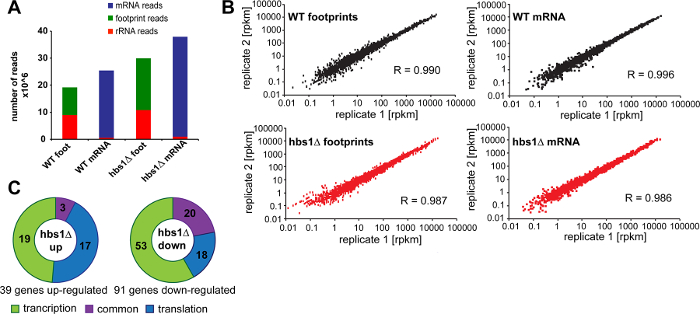
Figure 8: Representative results. (A) Number of mRNA, footprint, and rRNA reads obtained for wild-type sample and the hbs1Δ mutant. Combined number of reads generated by sequencing two biological replicates are shown for wild-type cells and the hbs1Δ mutant. (B) Reproducibility of footprint and mRNA-abundance measurements between two replicates. Pearson correlation coefficients (R) are indicated. (C) Transcriptional and translational changes in the hbs1Δ mutant. Significantly up-regulated and down-regulated genes in hbs1Δ are grouped in accordance to whether they are affected by a change in mRNA transcription, translation efficiency, or by a combined effect. Please click here to view a larger version of this figure.
| Primers | Sequence | Index | ||||
| 3' adapter (100 ng/µL) | /5rApp/AGATCGGAAGAGCACACGTCT/3ddC/ | |||||
| RT primer | pGATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGTCGCCGTATCATT/iSp18/GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||||
| Forward PCR primer | AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGACG | |||||
| Index primer 1 | CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | ATCACG | ||||
| Index primer 2 | CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | CGATGT | ||||
| Index primer 3 | CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | TTAGGC | ||||
| Index primer 4 | CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | TGACCA | ||||
| Index primer 5 | CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | ACAGTG | ||||
| Index primer 6 | CAAGCAGAAGACGGCATACGAGATATTGGCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | GCCAAT | ||||
Table 1: 3' Adapter and primer sequences.
