The GvEcR-based singular gene switch is depicted in Figure 1A. The pEUI(+) vector was optimized for regulated transgene expression by the treatment with Teb. The effector region of pEUI(+) comprises a 10xUAS and an E1b minimal promoter followed by an MCS containing EcoRI, PmlI, NheI, BmtI, AfeI, and AflII restriction enzyme recognition sites. An SV40 poly(A) signal was added behind the MCS (Figure 1B). A PAC gene was inserted between the driver and effector regions of pEUI(+) to confer resistance to puromycin.
To evaluate the activity and leakiness of pEUI(+), we subcloned EGFP into the EcoRI site of the MCS (pEUI(+)-EGFP), transiently transfected the plasmid construct into HEK293 cells, and administered Teb at different concentrations for 24 h (Figure 2). While mock vector transfectants did not show fluorescent signals regardless of Teb treatment (Figure 2A, B), pEUI(+)-EGFP transfectants became progressively sensitized to the increased amount of Teb (Figure 2C–F). More importantly, pEUI(+)-EGFP transfectants without treatment of Teb did not elicit any detectable EGFP signals under a fluorescent microscope (Figure 2C).
To further validate the responsiveness of pEUI(+) to Teb, we subcloned a FLAG epitope-tagged ANKRD13A gene into the EcoRI site of pEUI(+), transferred the construct into HEK293 cells, and then subjected them to puromycin selection for 3 weeks to establish a desired cell line with the ANKRD13A transgene. We analyzed the expression of the ANKRD13A transgene after a 24 h treatment with Teb and the established cell line responded well (Figure 3). We further demonstrated the reversibility of the pEUI(+) gene switch by depleting Teb from the cultural media. As shown in Figure 3, ANKRD13A expression was no longer detectable when we cultured the cells in Teb-free media.
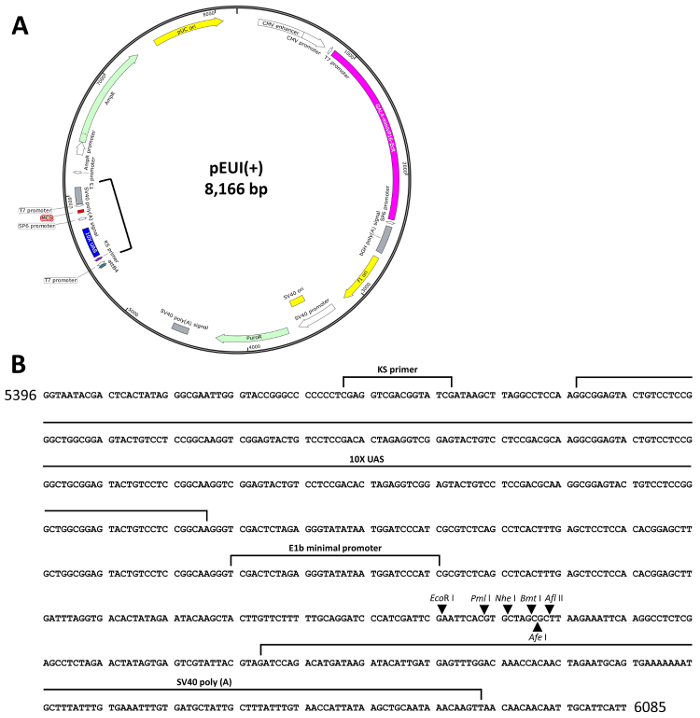
Figure 1. Schematic diagram of pEUI(+). (A) The vector map of pEUI(+) was constructed. A CMV promoter drives constitutive expression of GvEcR. The Teb-bound GvEcR will translocate into the nucleus and bind the 10xUAS cis-acting regulatory sequences to stimulate expression of the transgene inserted into the MCS. (B) The sequence information between the square brackets in (A) contains the entire effector sequences of pEUI(+). Arrows represent the individual restriction enzyme recognition sites included in the MCS. Please click here to view a larger version of this figure.
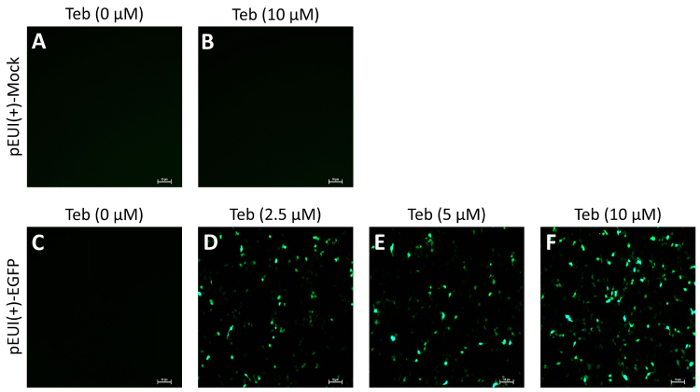
Figure 2. GvEcR-based singular gene switch responds to the treatment of Teb. HEK293 cells were transfected with the specified DNA constructs. After 24 h of transfection, the cells were stimulated for 24 h with Teb at the indicated concentration. EGFP transgene activity was observed under a fluorescent microscope. Mock transfectants with pEUI(+) did not show any detectable fluorescence with (A) or without (B) Teb treatment. (C–E) The fluorescent intensity in pEUI(+)-EGFP-transfected cells increases in a Teb dosage-dependent manner. Scale bar, 10 µm. Please click here to view a larger version of this figure.
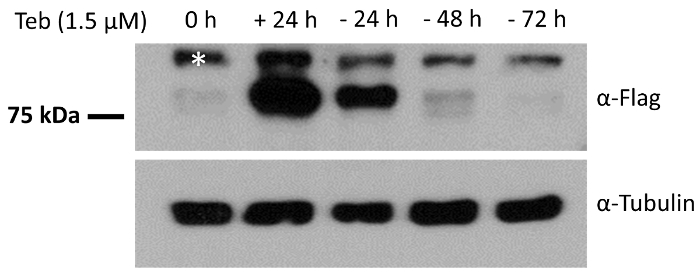
Figure 3. HEK293 cells stably harboring pEUI(+)/FLAG-ANKRD13A respond reversibly to the administration of Teb. FLAG epitope-tagged ANKRD13A expression was triggered by Teb (1.5 µM) treatment. After 24 h of treatment with Teb, the cells were switched to Teb drop-out media for the indicated amount of time. The relative amount of ANKRD13A was measured by Western blotting with anti-FLAG antibody. The endogenous expression level of α-tubulin was measured with anti-α-tubulin antibody as a control. The white asterisk indicates an unidentified cross-reactive species. Please click here to view a larger version of this figure.
