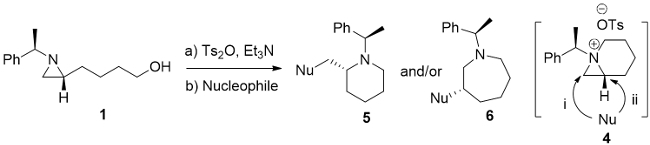
The reaction of 4-[(R)-1-(R)-1-phenylethyl)aziridin-2-yl]butan-1-ol (1)12 with p-toluenesulfonic anhydride and triethylamine in CH2Cl2 at room temperature for 1.0 h yielded the corresponding 2-(4-tosyloxybutyl)aziridine 2 in 96% yield11. 1H NMR (400 MHz) spectrum of compound 2 in CD3CN at different time intervals shows the conversion of 2-(4-tosyloxybutyl)aziridine 2 to 1-azabicyclo[4.1.0]heptane tosylate 4. Among all down field shifted peaks, a distinctive quartet peak at 2.34 ppm (J = 6.5 Hz) for tertiary proton of phenylethyl group was shifted to at 4.11 ppm (J = 6.9 Hz) by the formation of the quaternary ammonium ion (Figure 2).
The intermediacy of 1-azabicyclo[4.1.0]heptane tosylate 4 from 2-(4-tosyloxybutyl)aziridine 2 was further confirmed by the formation of azidated products, 2-azidomethylpiperidine 5a and 3-azidoazepane 6a, with sodium azide (NaN3) in CH3CN, while a simple reduction product 2-butylaziridine 3 was obtained from the reaction of the same starting substrate with lithium aluminium hydride (LiAlH4) in tetrahydrofuran (THF) (Figure 3).
The 1H and 13C NMR spectra of the ring expanded product 5a and 6a are depicted in Figure 5, Figure 6, Figure 7 and Figure 8, respectively. A distinguish quartet peak at 4.07 ppm with a coupling constant of 6.7 Hz in 1H NMR spectrum of compound 5a corresponds to the C-H proton of phenylethyl group present at nitrogen. Similar quartet at 3.81 ppm with a coupling constant of 6.8 Hz was observed for compound 6a. Similar NMR spectral changes were observed for all other compounds (Table 1). The detailed characterization data of 5a, 6a, 5f, 5h and 6j are reported as follows:
Compound 5a: Rf = 0.50 (hexanes/EtOAc 9:1 v/v). [α]20D = +64.3 (c = 0.6, CHCl3). 1H NMR (400 MHz, CD3CN) δ 7.43-7.17 (m, 5H), 4.07 (q, J = 6.7 Hz, 1H), 3.55 (dd, J = 12.7, 5.1 Hz, 1H), 3.47 (dd, J = 12.7, 6.8 Hz, 1H), 2.82-2.66 (m, 2H), 2.44-2.30 (m, 1H), 1.68-1.44 (m, 4H), 1.44-1.31 (m, 2H), 1.34 (d, J = 6.7 Hz, 3H). 13C NMR (CD3CN, 101 MHz): δ 144.8, 129.1, 128.5, 127.7, 59.0, 56.1, 50.6, 44.6, 28.1, 25.4, 21.8, 21.2. HRMS-MALDI (m/z): calcd. for C14H21N4 [M+H]+ 245.1760; found 245.1766.
Compound 6a: Rf = 0.70 (hexanes/EtOAc 9:1 v/v).[α]20D = -3.2 (c = 0.6, CHCl3). 1H NMR (400 MHz, CD3CN) δ 7.45-7.18 (m, 5H), 3.81 (q, J = 6.8 Hz, 1H), 3.49 (tt, J = 8.0, 5.1 Hz, 1H), 2.89 (dd, J = 13.9, 4.4 Hz, 1H), 2.64 (dd, J = 13.9, 7.7 Hz, 1H), 2.60-2.52 (m, 2H), 2.05-1.97 (m, 1H), 1.72-1.45 (m, 5H), 1.34 (d, J = 6.8 Hz, 3H). 13C NMR (CD3CN, 101 MHz): δ 145.5, 129.0, 128.4, 127.6, 64.5, 62.7, 56.9, 52.9, 33.5, 30.1, 22.7, 18.7. HRMS-MALDI (m/z): calcd. for C14H21N4 [M+H]+ 245.1760; found 245.1764.
2-[(R)-1-(R)-1-Phenylethyl)piperidin-2-yl]acetonitrile (5f): Rf = 0.85 (hexanes/EtOAc 19:1 v/v). [α]20D = +35.3 (c = 0.65, CHCl3). 1H NMR (400 MHz, CD3CN) δ 7.42-7.20 (m, 5H), 3.89 (q, J = 6.7 Hz, 1H), 2.85-2.75 (m, 2H), 2.68-2.54 (m, 2H), 2.45-2.37 (m, 1H), 1.71-1.44 (m, 5H), 1.42-1.31 (m, 1H), 1.34 (d, J = 6.7 Hz, 3H). 13C NMR (CD3CN, 101 MHz): δ 144.1, 129.2, 128.5, 127.9, 120.1, 59.3, 54.0, 44.3, 30.6, 26.1, 21.5, 21.2, 17.8. HRMS-MALDI (m/z): calcd. for C15H21N2 [M+H]+ 229.1699; found 229.1694.
[(R)-1-(R)-1-Phenylethyl)piperidin-2-yl)]methyl acetate (5h): Rf = 0.50 (hexanes/EtOAc 7:3 v/v). [α]20D = +75.8 (c = 0.55, CHCl3). 1H NMR (400 MHz, CD3CN) δ 7.41-7.26 (m, 4H), 7.25-7.17 (m, 1H), 4.29 (dd, J = 11.3, 4.8 Hz, 1H), 4.16 (dd, J = 11.3, 6.7 Hz, 1H), 4.05 (q, J = 6.8 Hz, 1H), 2.81-2.68 (m, 2H), 2.40 (ddd, J = 12.1, 6.2, 3.4 Hz, 1H), 1.97 (s, 3H), 1.65-1.41 (m, 5H), 1.39-1.29 (m, 1H), 1.32 (d, J = 6.8 Hz, 3H). 13C NMR (CD3CN, 101 MHz): δ 171.5, 145.2, 129.0, 128.5, 127.6, 63.8, 59.4, 55.7, 45.3, 28.8, 26.3, 22.1, 21.1, 21.0. HRMS-MALDI (m/z): calcd. for C16H24NO2 [M+H]+ 262.1801; found 262.1800.
(S)-1-[(R)-1-Phenylethyl]azepan-3-ol (6j): Rf = 0.40 (hexanes/EtOAc 1:1 v/v). [α]20D = -11.1 (c = 0.55, CHCl3). 1H NMR (400 MHz, CD3CN) δ 7.41-7.27 (m, 4H), 7.27-7.19 (m, 1H), 3.80 (q, J = 6.8 Hz, 1H), 3.67-3.56 (m, 1H), 2.71 (dd, J = 13.5, 3.1 Hz, 1H), 2.67-2.53 (m, 3H), 1.72-1.43 (m, 6H), 1.35 (d, J = 6.8 Hz, 3H). 13C NMR (CD3CN, 101 MHz): δ 145.4, 129.0, 128.6, 127.7, 70.2, 64.4, 57.1, 53.1, 37.9, 29.7, 21.7, 17.6. HRMS-MALDI (m/z): calcd. for C14H22NO [M+H]+ 220.1695; found 220.1693.
The aziridine-ring-expansion has been observed with several other nucleophiles and the results are summarised in Table 1. Among all nucleophiles, cyanide, thiocyanide and acetate preferred the selective formation of piperidine rings (Table 1, entry f, g, h), while azide, hydroxide and amine nucleophiles yielded azepane rings (Table 1, entry a, j, k).
The structures and their stereochemistry of major compounds 5f, 5h and 6j originatd from (2R)-2-(4-hydroxybutyl)-1-[(1R)-(1-phenylethyl)]aziridine were further confirmed by their conversion to known compounds13,14,15 and we found out that the nucleophilic ring-expansion proceeds in completely stereospecific manner. Representative examples include 2-azidomethyl- (5a), 3-azidoazopane (6a), 2-cyanomethyl-(5f), 2-acetyloxymethylpiperidine (5h) and 3-hydroxyazepane (6j) in their optically pure froms (Figure 4).
The presented protocol was applicable to synthesize natural azasugar D-Fagomine (9) and its 3-epimer16. The reaction of suitably functionalized aziridinyl alcohol 7 underwent aziridine ring-expansion under similar reaction condition to afford protected fagomine 8. The removal of all protecting group in compound 8 produces D-fagomine (9) in 94% yields (Figure 9)11.
In the same manner, benzyl protected alcohol 10 was treated with p-toluenesulfonic anhydride and Et3N, followed by nucleophilic ring opening with sodium cyanide (NaCN), resulting in the ring expanded cyanomethylpiperidine 11 in 90% yield as a single isomer, which can be used as synthetic precursor for febrifugine analogue (Figure 10)17.
The ring-expansion toward azepane also succeeded via azoniabicycle prepared from alcohol 13 followed by the reaction with NaN3 to yield the desired product 14, which was used for azepane core of balanol (15) by one-pot benzyl deprotecton and azide reduction under catalytic hydrogenation (Figure 11)11.
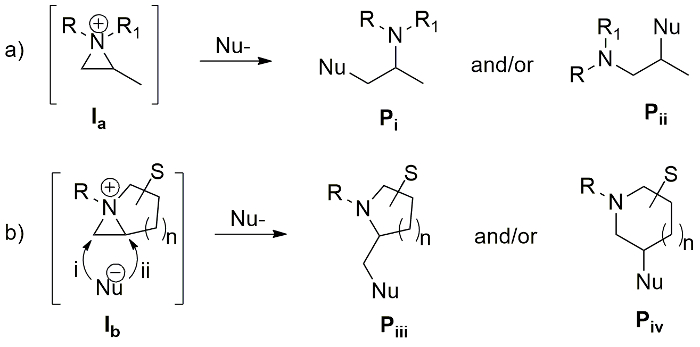
Figure 1. Ring opening of aziridinium ion. Possible ring opening products: (a) Pi and Pii from monocyclic aziridinium ion (Ia); (b) Piii and Piv from bicyclic aziridinium ions (Ib). Please click here to view a larger version of this figure.
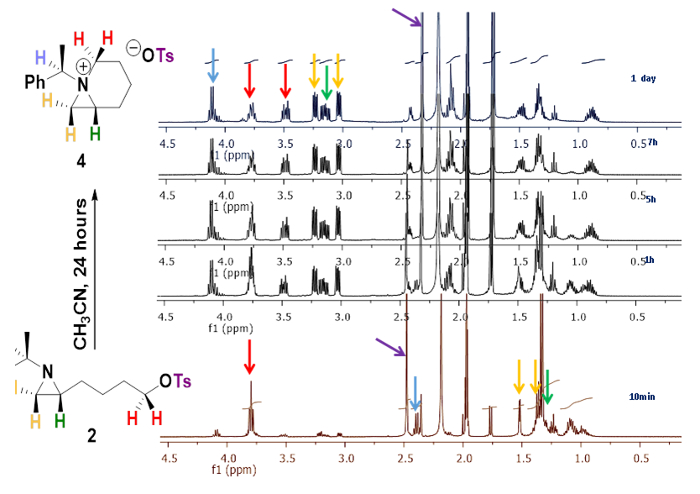
Figure 2. Generation of bicyclic aziridinium ion. 1H NMR (400 MHz) spectral changes by the conversion of 2-(4-tosyloxybutyl)aziridine 2 to 1-azoniabicyclo[4.1.0]heptane tosylate 4 in CD3CN in different time intervals as 10 min, 1, 5, 7 and 24 h11. Please click here to view a larger version of this figure.
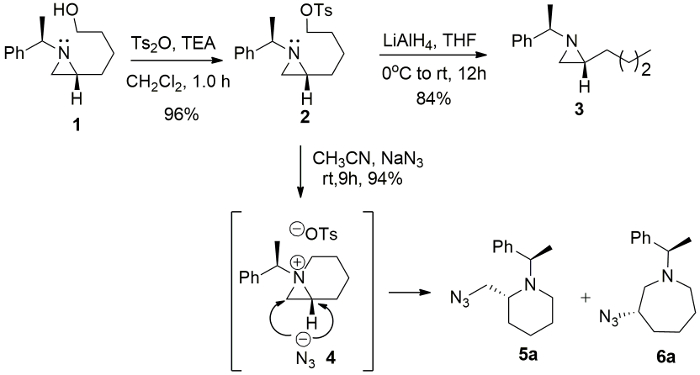
Figure 3. Ring expansion of aziridine. The formation of 1-azoniabicyclo-[4.1.0]heptane tosylate 4 and its ring-expansion reaction with NaN3. Please click here to view a larger version of this figure.
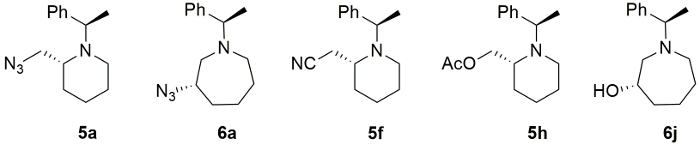
Figure 4. Piperidines and azepanes products. Representative ring expansion products (5a, 6a, 5f, 5h and 6j) Please click here to view a larger version of this figure.
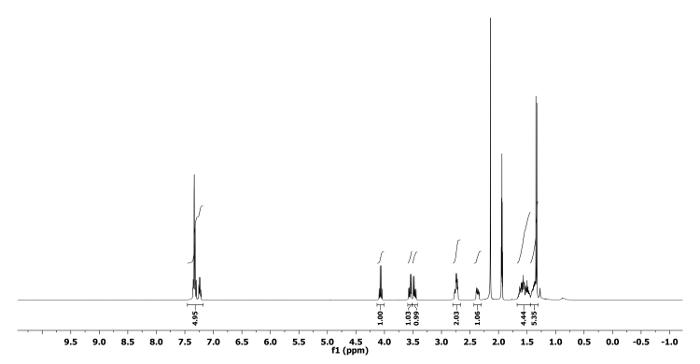
Figure 5. 1H NMR spectrum of 5a. Chemical shifts and relative integrations of characteristic protons are shown11. Please click here to view a larger version of this figure.
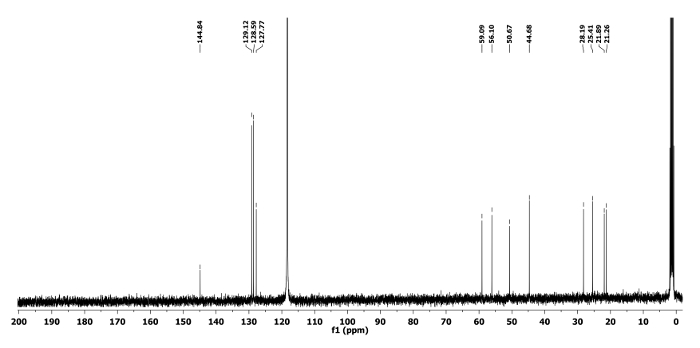
Figure 6. 13C NMR spectrum of 5a. Chemical shifts of characteristic carbons are shown11. Please click here to view a larger version of this figure.
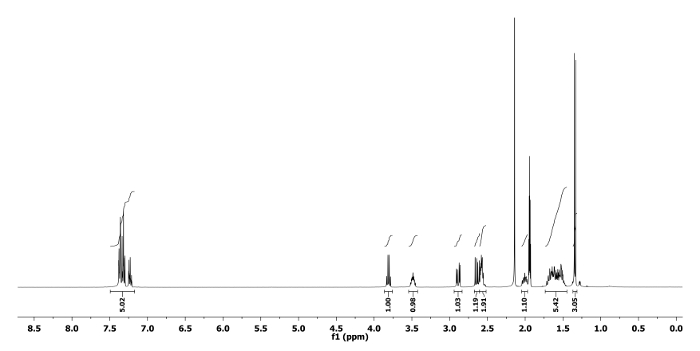
Figure 7. 1H NMR spectrum of 6a. Chemical shifts and relative integrations of characteristic protons are shown11. Please click here to view a larger version of this figure.
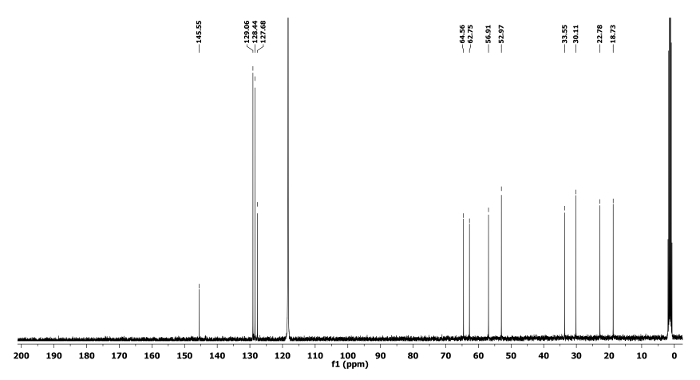
Figure 8. 13C NMR spectrum of 6a. Chemical shifts of characteristic carbons are shown11. Please click here to view a larger version of this figure.
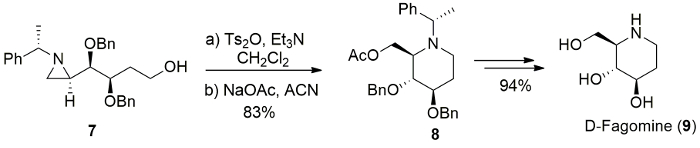
Figure 9. Synthesis of fagomine. Synthesis of fagomine (9) from the ring-expansion of (3R,4R)-3,4-bis(benzyloxy)-4-[(R)-1-(S)-1-phenylethyl)aziridin-2-yl]butan-1-ol (7). Please click here to view a larger version of this figure.
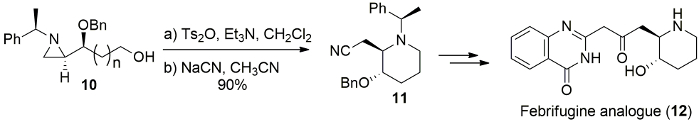
Figure 10. Synthesis of febrifugine. Synthesis of febrifugine analogue (12) from the ring-expansion of (S)-4-(benzyloxy)-4-[(R)-1-(R)-1-phenylethyl)aziridin-2-yl]butan-1-ol (10). Please click here to view a larger version of this figure.
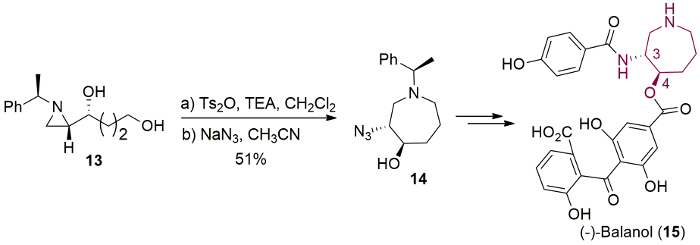
Figure 11. Synthesis of azepane natural product. Synthesis of (-)-balanol (15) from the ring-expansion of (R)-1-[(S)-1-(R)-1-phenylethyl)aziridin-2-yl]butane-1,4-diol (13) as a key step. Please click here to view a larger version of this figure.
 |
||||||
| Entry | Substrate | Nucleophile | Solventa,b | Time (h) | Ratio (5/6)c | Yieldd (%) |
| a | Aziridine (1) | NaN3 | CH3CN | 9 | 41/59 | 94 |
| b | Aziridine (1) | NaN3 | 1,4-dioxane | 2.0 | 40/60 | 84 |
| c | Aziridine (1) | CsF | CH3CN | 8.0 | 46/54 | 79 |
| d | Aziridine (1) | n-Bu4NBr | CH3CN | 12.0 | Complex | – |
| e | Aziridine (1) | NaI | CH3CN | 12.0 | Complex | – |
| f | Aziridine (1) | NaCN | CH3CN | 8.0 | 92/08 | 92 |
| g | Aziridine (1) | NaSCN | CH3CN | 11.0 | 91/09 | 93 |
| h | Aziridine (1) | NaOAc | CH3CN | 12.0 | 60/40 | 90 |
| i | Aziridine (1) | NaOMe | CH3CN | 11.0 | 58/42 | 93 |
| j | Aziridine (1) | NaOH | 1,4-dioxane | 2.0 | 15/85 | 93 |
| k | Aziridine (1) | BnNH2 | CH3CN | 14.0 | 35/65 | 87 |
| l | Aziridine (1) | Phenol | CH3CN | 15.0 | 47/53 | 56 |
| m | Aziridine (1) | NaOBz | CH3CN | 13.0 | 48/52 | 76 |
| n | Aziridine (1a) | NaN3 | CH3CN | 9.5 | 40/60 | 91e |
| [a]CH2Cl2 was used as solvents for the preparation of 2-(4-tosyloxybutyl)aziridine. [b]Tosylation was performed at 0 to 25 °C. | ||||||
| [c]Determined by 1H NMR. [d]Combined yield of 5 and 6. [e]Inseparable mixture of 5n and 6n from isomeric aziridine 1a. | ||||||
| [f] All reactions were performed at 25 °C except the entries b and j (110 °C). [g]Concentration is 0.1 M | ||||||
Table 1. Selected examples of aziridine ring expansion. The formation and ring-expansion of 1-azoniabicyclo[4.1.0]heptane tosylate 4 with various nucleophiles11.
