In a successful experiment, the secondary antibody will specifically pinpoint the location of the specific epitope in bright green, in a consistent manner, allowing for the characterization of the cell wall composition at a certain development stage of the cell, tissue or organ. For example the LM6 antibody has an high affinity for 1,5-arabinan, a compound with type-I rhamnogalacturonan that can be found abundantly labelling the cell wall of the developing Quercus suber anther (Figure 2A), thus allowing to conclude that this type of pectin is abundant and part of the primary cell wall composition. JIM5 has affinity for homogalacturonans scarcely esterified that are typically found at the root tip of Quercus suber embryo, specifying mechanical properties of the organ (Figure 2B). Xylogalacturonan are a type of pectin rich in xylose associated with cell wall loosening, they are found in degenerating cells. The antibody LM8 specifically recognizes xylogalacturonans, in maturing organs it may be used to detect degenerating cells or tissues, like the endosperm cells during the final stages of the Quercus suber acorn maturation (Figure 2C).
JIM13 has affinity for AGPs found on structures related with reproduction, cell lines related to microgametogenesis in Arabidopsis thaliana (Figure 2D). JIM8 also recognizes epitopes of AGPs present in cells and organs related to reproduction like the stigmatic papillae and micropyle of the Basal Angiosperm Trithuria submersa (Figure 2E-2F). Both antibodies have been suggested to be molecular markers for the gametophytic cell lines in plants9.
Common mistakes to this protocol are normally easy to detect and identify. When the washes are skipped or the reaction wells are let to dry, the secondary antibody will usually appear as an unspecific smear covering indiscriminately over cells, tissues, resin and slide (Figure 3A). Aggregates of green fluorochrome will form if the unbounded primary antibody has not been properly washed away (Figure 3B). Another common cause for failure of this technique is related with the folding and/or detachment of the sections (Figure 3C), rendering the experiment useless. This problem is usually related with either poor adhesion of the sections to the slides, probably due to the use of unclean slides, and/or aggressive washing.
The sample preparation is also a critical step in this procedure. Fortunately, the most common fixative and embedding issues are easy to spot (Figure 3D). If all goes well the resin block will be free of cracks and the sample will be clearly visible with a pale yellow to light brown color (Figure 3D-1). Samples inefficiently embedded will show powdery white spots or areas (Figure 3D-2). Keeping the sample size under 8 mm is important to guarantee the penetration of the fixative solution. When samples are poorly fixed, they will appear dark brown almost black (Figure 3D-3). Also the temperature of polymerization is important for both the preservation of the epitopes and proper hardening of the resin. An excessive temperature can cause the resin to crack making the sectioning of the sample impossible (Figure 3D–4).

Figure 1. Overview of the complete protocol. Please click here to view a larger version of this figure.
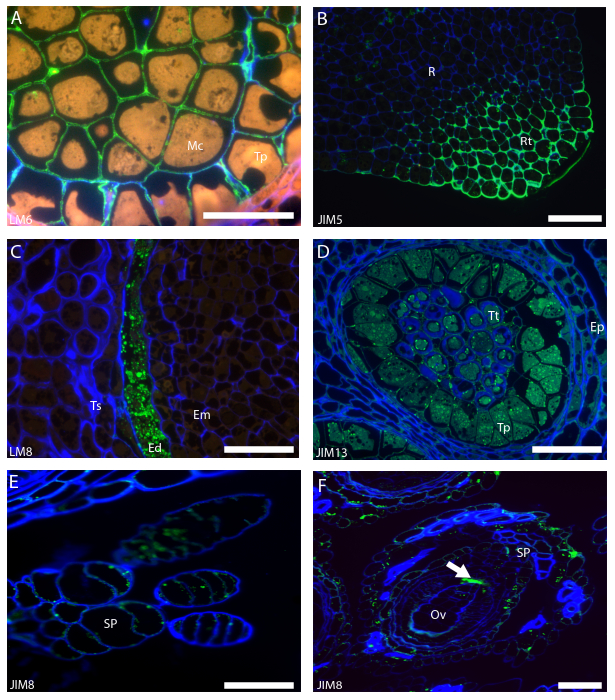
Figure 2: Typical results of AGPs and Pectin immunolocalization in plant samples. (A) LM6 labeling of arabinan moiety of pectins in a Quercus suber anther in meiosis I. (B) Specific labeling of low methyl-esterified pectins in a Quercus suber embryo root tip by JIM5. (C) Xylogalacturonan labeled by LM8 in the receding endosperm of a Quercus suber maturing acorn. (D) JIM13 labeling of AGPs in the tapetum and tetrads of an Arabidopsis thaliana anther. (E) AGPs labeled by JIM8 in the stigmatic papillae of Trithuria submersa. (F) JIM8 labelling AGPs at the micropyle (white arrow) of a Trithuria submersa ovule. Meiotic microspore (Mc), Tapetum (Tp), Root (R), Root tip (Rt), Testa (Ts), Endosperm (Ed), Embryo (Em), Epidermis (Ep), Stigmatic papillae (SP), Ovule (OV). Scale bars: 100 µm. Please click here to view a larger version of this figure.
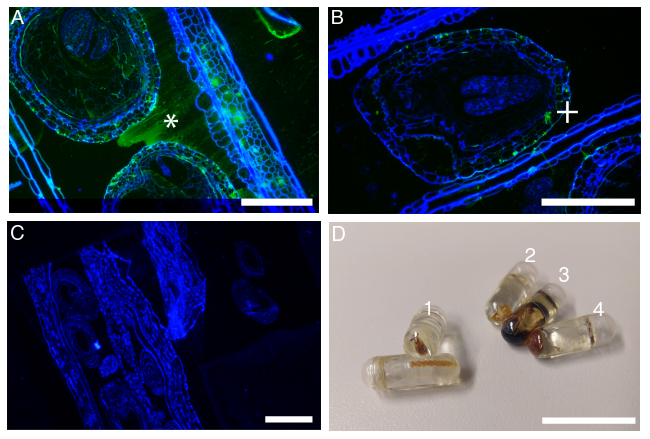
Figure 3: Common mistakes and problems during the protocol. (A) Failure of wash steps are identified by the presence of unspecific smear of secondary antibody (+) over sample resin. (B) The poor specificity or wash failure of the primary antibody results in the formation of FITC aggregates (+) not bonded to a specific location. (C) Folds and section detachment are often the result of aggressive wash and unclean slides. (D) Examples of sample fixation and embedding; (1) perfect sample size and embedding, (2) embedding failure, (3) sample fixation failure, (4) resin hardening failure. FITC labeled antibody (green) and Calcofluor-white stain (blue). Scale bars: 100 µm. Please click here to view a larger version of this figure.
Supplemental Figure 1. (A) Schematic representation of the block trimming sequence for preparing the sample (Yellow block) for sectioning with the ultramicrotome. Firstly, the gelatin capsule is removed (1). Then they are first trimmed at a 40° to 45° angle to the sides of the capsule tangentially to the sample (2), a second cut is made at 90° of the first (3) followed by a third (4) and fourth following the same rule (5). From this first phase of trimming results a pyramidal shaped structure which summit is located above the sample. Finally, with a sharp blade the summit is shaved off perpendicularly to the resin block major axis to reach the embedded sample. A square or trapezoid surface should be obtained at the end of the procedure (6). (B) Required supplies to make an incubation chamber; tin foil (1), double sided duct tape (2), paper towels (3) and a pipette tip box. (C) First step, the tin foil is fixed to the box with the double-sided duct tape. (D) Second step, the pipette tips holder is removed to place paper towels at the bottom of the box. (E) Third step, the paper towels are damped with water and the pipette tips holder is placed back to act has a tray for the slides. Please click here to download this file.
Supplemental Table 1: List of useful monoclonal antibodies. The above table represents an example of available antibodies, with information about their targets and where they can be purchased. Please click here to download this file.
