Sinapinic acid was synthesized in high purity and high yield (> 95%) from syringaldehyde using the Green Knoevenagel condensation. (Supporting Information: Figure S1) The E-factor is an indication of waste production where a higher number indicates more waste. The E-factor is calculated by taking the total material input, subtracting the amount of the desired end product, and dividing the whole by the amount of the end product. This Green Knoevenagel condensation has an E-factor of 1.0, which can be calculated: [((29.81 g malonic acid + 36.4 g syringaldehyde + 0.790 g ammonium bicarbonate + 18.04 g ethyl acetate) – 42.56 g sinapinic acid) / 42.56 g sinapinic acid]. The sinapinic acid was hydrogenated with a high yield (>95%) to obtain the intended monomer dihydrosinapinic acid. (Supporting Information: Figure S2) This hydrogenation reaction has an E-factor of 0.84. Subsequently, dihydrosinapinic acid was acetylated to obtain 4-acetoxy-dihydrosinapinic acid and its prepolymer. The E-factor of this acetylation reaction was 0.45, assuming full conversion to 4-acetoxy-dihydrosinapinic acid (Supporting Information: Figure S3).
Usually, the E-factor of organic synthetic reactions of pharmaceuticals is between 5 and 100, and these reactions frequently use relatively harmful chemicals21. The E-factor, calculated for both the Green Knoevenagel and subsequent reactions, is remarkably low compared to these numbers. Yet, the E-factor exhibits some shortcomings. For example, the environmental hazard of a given waste component is not reflected by the E-factor. In principle, this can be compensated by the "environmental quotient" (Q) taking the "environmental unfriendliness" of a given waste into account. Because the catalyst ammonium bicarbonate is relatively harmless, the environmental unfriendliness quotient Q of the Green Knoevenagel is also stated as low. Still, additional research is essential to confirm this22. By applying the mentioned organic synthetic reactions, 4-acetoxy-dihydrosinapinic acid and its prepolymers are ready for the polycondensation to poly-S.
Figure 1 depicts the Proposed mechanism of the step-growth polymerization of 4-acetoxy-dihydrosinapinic acid. In step i, the added base starts the reaction and controls the reaction rate by deprotonating the acid group, thereby stimulating its reactivity23. The formed carboxylate attacks the carbonyl of the acetoxy group and creates a tetrahedral. In this state, the phenoxide group connects to the other carbonyl through a phenoxide shift, which was initially the aliphatic carboxylic acid group. When the phenoxide shift has occurred, the acetate-ion leaves the intermediate, subtracts a proton from the original base and then leaves the reaction as acetic acid. In practice, this step is promoted by creating a vacuum in the reaction vessel24. In each cycle, one novel molecule is added to the chain. During the progress of the polycondensation reaction, it turned out to be challenging to find the exact combination between increasing the molecular weight and degradation of the polymer chains. As the temperature was elevated with a gradient during the melt polymerization, growing amounts of degradation occurred as the viscosity of the polymer increased. A small amount of solvent was added during the second step of the polycondensation to decrease the viscosity. In this solvent-assisted poly-condensation, 1,2-xylene turns out to be a suitable agent for promoting the polycondensation25.
Figure 2 represents three different 1H NMR measurements during the polymerization process. The samples dissolved well and were measured in a mixture of CF3COOD / CDCl3. The 1H NMR spectra were taken after the acetylation of sinapinic acid. The 1H NMR spectrum in the middle is a sample taken from the same reaction. No specific compound was isolated, so acetylated sinapic acid and its prepolymers are shown. The 1H NMR spectrum above was taken from a sample after completion of the polycondensation. The areas of the acetyl end groups decrease during the polymerization process, and the number average degree of polymerization (DPn) increases. After completing the reaction, the degree of polymerization was set at 43 repeating units as determined by 1H NMR measurements.
GPC measurements were performed to investigate the chain length of poly-S, and various GPC-systems have been tested for the analysis. The initial step was to dissolve the polymeric material in several common organic solvents such as chloroform (CHCl3), dimethylformamide (DMF), hexafluoro-2-propanol (HFIP), and tetrahydrofuran (THF). However, it appears that the highly crystalline material of poly-S, visible on DSC thermograms, was sparingly soluble in these solvents. Also, when the polymeric material was melted and then cooled very quickly in liquid nitrogen to increase the amorphicity of the polymer, only the short polymer chains dissolved in DMF. The value measured on GPC was only 1,900 up to 2,100 Da, which corresponds to approximately ten repeating units.
Another pretreatment method has been selected to perform a proper GPC measurement. Additional research26 prescribes that a more amorphous polyester material can be formed when polystyrene in its atactic form is melt-mixed into the polyester. The two polymers (polyester and polystyrene) were placed together in a DSC pan, melted, and subsequently cooled in liquid nitrogen. Washing the quenched mixture with acetone removed the residual polystyrene, and after successfully dissolving in DMF, a high molecular weight polymer could be observed.
When poly-S was hydrolyzed in 1 M NaOH, the molecularly recyclable polyester yielded the starting material dihydrosinapinic acid in less than 10 minutes. HPLC, melting point analysis, and 1H NMR confirmed this observation (Supporting information: Figure S4). Extended reaction times did not increase the yield, as shown in Figure 3.
The thermograms and GPC analysis of poly-S 1.0 and their successive generations poly-S 2.0 and poly-S 3.0 after forced amorphicity with atactic polystyrene are shown in Figure 4 and Table 1, respectively. DSC analysis of poly-S shows a glass transition signal (Tg) at 113 °C and an endothermic melting signal (Tm) at 281 °C. The polymers from re-polymerized sinapinic acid 3a poly-S 2.0 up and poly-H 3.0 exhibit similar thermal properties (Supporting Information: Figures S5-S7). Poly-S is a semi-crystalline polymeric material because glass transition temperatures (Tg) and melting signals are present in the thermograms of poly-S and their successive generations. It can be stated GPC analysis after forced amorphicity with atactic polystyrene displays a constant length of polymeric material throughout the different generations (Supporting Information: Figures S8-S10).
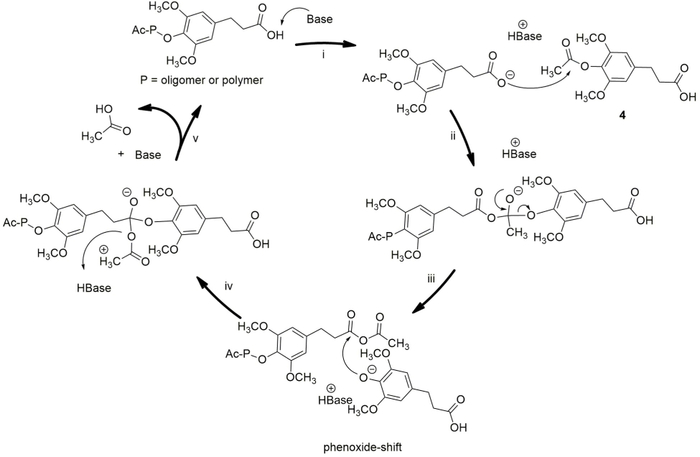
Figure 1: Proposed mechanism of the step-growth polycondensation of 4-acetoxy-dihydrosinapinic acid. Please click here to view a larger version of this figure.
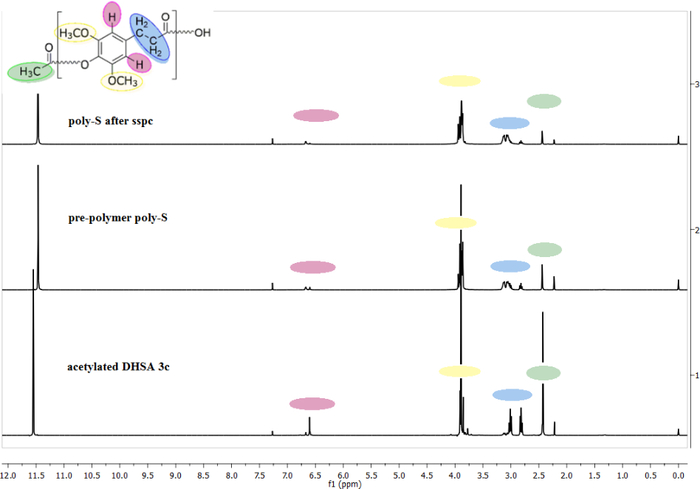
Figure 2: Overlay of 1H NMR spectra (25 °C, CF3COOD/CDCl3, with residual solvent peaks at 11.49 and 7.27 ppm for CF3COOH and CHCl3, respectively) of the isolated acetylation product of sinapinic acid (bottom); the prepolymer of poly-S (middle); poly-S after solid-state post condensation (240 °C, 30 min, vacuum) (top). Please click here to view a larger version of this figure.
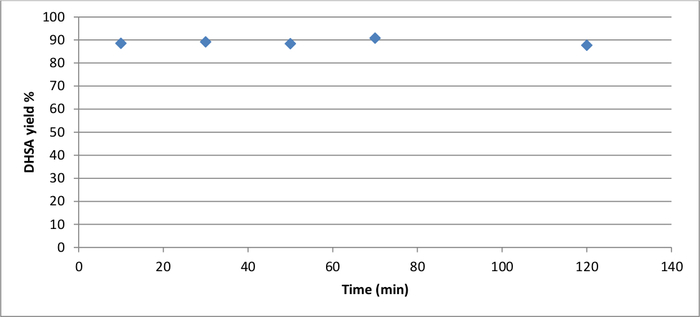
Figure 3: Alkaline hydrolysis (1 M NaOH solution, 80 °C) of poly-S over time, determined by dihydrosinapinic acid (DHSA) yield based on HPLC peak areas. Please click here to view a larger version of this figure.
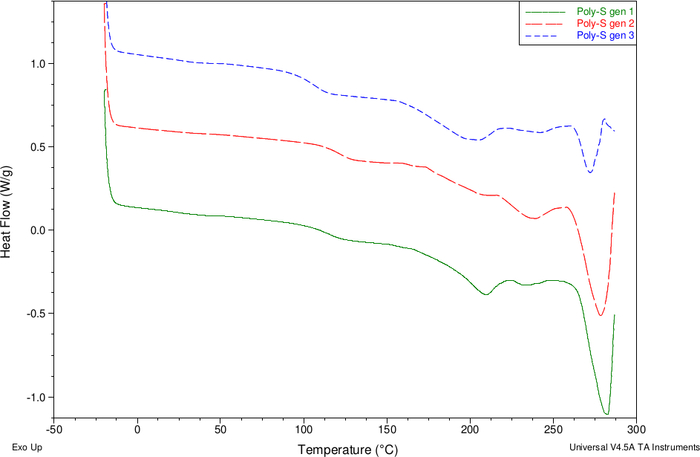
Figure 4: DSC thermogram of three generations poly-S. Please click here to view a larger version of this figure.
| Generation | Poly-S (mg) | PS (mg) | Mn | Mw | PDI | DP |
| 1.0 | 5.0 | 4.8 | 120030 | 193580 | 1.6 | 594 |
| 2.0 | 5.0 | 5.0 | 134740 | 194410 | 1.4 | 667 |
| 3.0 | 4.7 | 4.9 | 153620 | 237210 | 1.5 | 760 |
Table 1: GPC measurements of poly-S generations.
