Protein post-translational modifications (PTMs) play a major role in modulating protein structures and consequently their functions and downstream biological processes. The diversity of the human proteome increases exponentially due to the combinatorial variability afforded by various PTMs. Different variants of proteins from their canonical sequences as predicted by the genome are known as proteoforms, and many proteoforms arise from PTMs1. Studying proteoform diversity in health and disease has become an area of research of great interest in recent years2,3.
The study of proteoforms and more specifically PTMs with great depth has become more facile through the development of mass spectrometry (MS)-based proteomics methods. Using MS, analytes are ionized, fragmented, and identified based on the m/z of fragments. Enrichment methods are often necessary due to the low relative abundance of PTMs compared to non-modified forms of proteins. Though analysis of intact proteins and their PTMs, called top-down analyses, have become more routine, the enzymatic digestion of proteins and the analysis of their component peptides in bottom-up analyses is still the most widely used route for PTM analysis. The two most widely studied PTMs, and the two most common PTMs in vivo, are glycosylation and phosphorylation4. These two PTMs play major roles in cell signaling and recognition and thus are important modifications to characterize in disease research.
The chemical properties of various PTMs often provides routes toward enrichment of these PTMs at the protein and peptide levels prior to analysis. Glycosylation is a hydrophilic PTM due to the abundance of hydroxyl groups on each monosaccharide. This property can be used to enrich glycopeptides in hydrophilic interaction chromatography (HILIC), which can separate more hydrophilic glycopeptides from the hydrophobic non-modified peptides5. Phosphorylation adds the phosphate moiety, which is negatively charged except at acidic pH. Due to this charge, various metal cations, including titanium, can be used to attract and bind phosphopeptides while non-phosphorylated species are washed away. This is the principle of immobilized metal affinity chromatography (IMAC). Further discussions of these and other enrichment strategies for glycosylation and phosphorylation can be found in recent reviews6,7.
Comparatively large amounts of starting peptide material (0.5 mg or more) are often needed for enrichment protocols due to the low stoichiometry of PTMs on peptides. In scenarios where this amount of sample may not be easily obtained, such as tumor core biopsy or cerebrospinal fluid analyses, it is beneficial to use facile workflows that result in maximum biomolecular information. Recent strategies developed by our lab and others have highlighted the simultaneous and parallel analysis of glycosylation and phosphorylation using the same PTM enrichment workflow8,9,10,11,12. Though the chemical properties of these two PTMs may differ, these PTMs may be analyzed in multiple steps due to the innovative separation techniques and materials used. For example, electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) overlays separations based on hydrophilic interactions between analytes and the mobile phase with charge-charge interactions between analytes and the stationary phase material13,14,15,16. At acidic pH, the attraction of phosphorylated peptides to the stationary phase can improve their retention and separation from non-modified peptides. Material consisting of Ti(IV) immobilized on hydrophilic microspheres can be used for HILIC and IMAC-based elution to separate phosphopeptides and neutral, acidic, and mannose-6-phosphorylated glycopeptides17,18. This strategy is known as dual-functional Ti(IV)-IMAC. Using these strategies for enriching multiple PTMs in a single workflow can make analyses of potential PTM crosstalk interactions more accessible. Additionally, the total sample amount and time requirements are less than the conventional enrichment methods when performed in parallel (i.e., HILIC and IMAC on separate sample aliquots).
To demonstrate the dual-functional Ti(IV)-IMAC strategy for simultaneous analysis of protein glycosylation and phosphorylation, we have applied it to analyze post-mortem human pancreatic tissues. The pancreas produces both digestive enzymes and regulatory hormones, including insulin and glucagon. The pancreatic function is impaired in pancreatic disease. In diabetes, the regulation of blood sugar is affected, leading to higher levels of glucose in the blood. In pancreatitis, inflammation results from auto-digestion of the organ3. Changes in PTM profiles, including glycosylation and phosphorylation, may result, as is often the case, in other diseases.
Here, we describe a protocol for a spin-tip based simultaneous enrichment method, based on a dual-functional Ti(IV)-IMAC strategy, for N-glycopeptides and phosphopeptides derived from proteins extracted from pancreatic tissue. The protocol includes protein extraction and digestion, enrichment, MS data collection, and data processing, as can be seen in Figure 1. Representative data from this study are available via ProteomeXchange Consortium with identifier PXD033065.
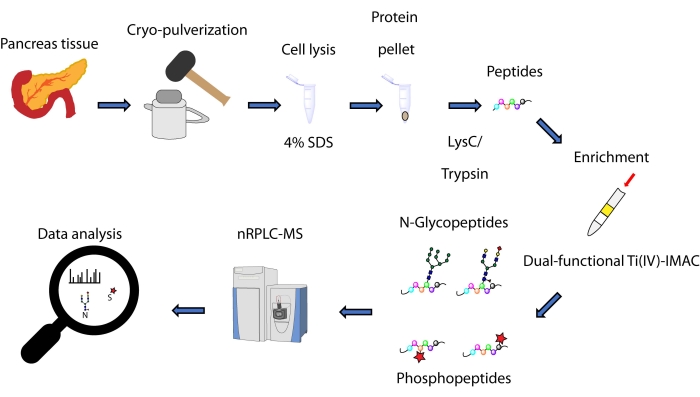
Figure 1: Workflow for simultaneous analysis of N-glycopeptides and phosphopeptides from human pancreatic tissues. Tissues are first cryo-pulverized into a fine powder before protein extraction using the detergent sodium dodecyl sulfate (SDS). Proteins are then subjected to enzymatic digestion. The resulting peptides are aliquoted prior to enrichment using dual-functional Ti(IV)-IMAC. Raw data is collected using nanoscale reversed phase liquid chromatography-mass spectrometry (nRPLC-MS) and is analyzed using database searching software. Please click here to view a larger version of this figure.
This protocol is intended to make PTM analyses more accessible and to enable more widespread analysis of multiple PTMs in the same workflow. This protocol can be applied to other complex biological matrices, including cells and biofluids.
Representative mass spectrometry data, including raw files and search results, have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD03306522.
In this work, duplicate injection replicates were analyzed for each enrichment elution. Identifications made from both technical replicates were collated in the final analysis. Due to the semi-stochastic nature of data-dependent acquisition in picking peptide precursors for MS/MS fragmentation, identification overlap of about 70%-80% is expected between technical duplicates. In applications using these methods, at least two technical replicates are encouraged. For downstream statistical analyses, biological replicates for different experimental conditions should be taken into consideration. Depending on the desired stringency for data reporting, it may be useful to set filters for identifications, e.g., identification in at least x number of technical or biological replicates to be reported.
An example total ion chromatogram (TIC) for each elution from each enrichment can be seen in Supplementary Figure S1, Supplementary Figure S2, and Supplementary Figure S3. Using stringent filters during database searching of raw MS files can ensure more accurate and more confident identifications of MS/MS spectra to peptide sequences with PTMs. As is evident in the TICs for each elution, the peptide samples are still complex even after fractionation from the enrichment. Nanoflow chromatography coupled to a high-resolution, high-mass accuracy, and fast-scanning mass spectrometer can help analyze complex mixtures, such as peptides from a tryptic digest, with great sensitivity and depth. Example MS/MS spectra for glycosylated and phosphorylated spectra can be seen in Figure 2. Well-annotated spectra such as these result from rich fragmentation of precursors which increase confidence in the identification as assigned by the database searching software.
Figure 3 illustrates the peptide identifications made using the dual-functional Ti(IV)-IMAC spin-tip enrichment method over each elution fraction. The majority of glycopeptides elute in the first four fractions, while the majority of phosphopeptides elute in the last three fractions. This separation of different PTMs among the fractions helps prevent any interference in ionization that may result if they were not as adequately separated across elutions.
Figure 4 compares the glycoproteomics results from the dual Ti method compared to an ERLIC glycopeptide-only enrichment. As can be seen in Figure 4A, many identifications at the protein, glycoform, PTM, and modification site levels are common to both methods. ERLIC has more unique identifications at all levels compared to the dual Ti method, however. This includes M6P (high mannose glycans with at least one phosphate group) glycoforms, which require additional manual curation of the data to remove false positive identifications. Further, the types of glycans identified using either method are similar. The proportions of each type of glycan after binning into six categories based on their compositions are similar between both methods16.
Phosphoproteomics results from the dual Ti method are compared to a conventional IMAC phosphopeptide-only enrichment in Figure 5. The dual-functional Ti(IV) enrichment performs similarly to conventional IMAC as shown by substantial overlap at the protein, peptide, and PTM levels. Using either method, the majority of phosphorylated amino acids are serine and threonine, with around 1% phosphorylated tyrosine identified as well. Both the methods can also be used to identify multi-phosphorylated peptides.
The lists of modified proteins are mapped to genes and analyzed using gene ontology (GO) enrichment as shown in Figure 6 and Figure 7. GO analyses for the modified proteins identified using ERLIC and IMAC are shown in Supplementary Figure S4 and Supplementary Figure S5. The free, online Metascape tool performs the enrichment and creates networks based on enriched pathways and processes, linking them by similarity23. The node terms for each GO analysis can be found in Supplementary Table S1, Supplementary Table S2, Supplementary Table S3, and Supplementary Table S4. Since glycosylation is a major protein PTM in the extracellular matrix, it is not surprising that several terms enriched using the list of N-glycoproteins identified are related to the extracellular matrix. Phosphorylation is involved in cell signaling and this can be seen in several enriched terms found using the list of phosphoproteins identified.
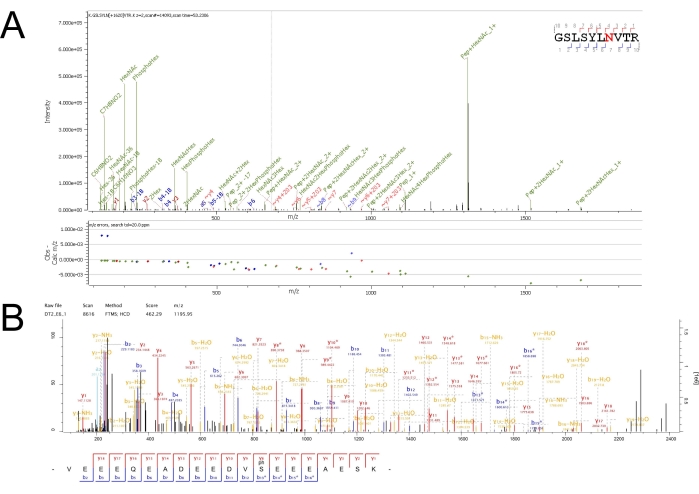
Figure 2: Annotated high confidence MS/MS spectra from N-glycosylated and phosphorylated peptides. (A) Mannose-6-phosphorylated peptide GSLSYLN(HexNAc(2)Hex(7)Phospho(1))VTR from cathepsin D (CATD) identified from retention times 53.08-53.33 min from the seventh enrichment elution. The presence of the PhosphoHex oxonium ion at 243 m/z improves confidence in this PTM assignment. (B) Phosphorylated peptide VEEEQEADEEDVS(Phospho)EEEAESK from thioredoxin-related transmembrane protein 1 (TMX1) from retention times 32.31-32.72 min from the sixth enrichment elution. The presence of -98 (-H3PO4) neutral losses between peptide fragment ions and their de-phosphorylated variants (denoted by fragment*) improves confidence in this PTM assignment. Please click here to view a larger version of this figure.
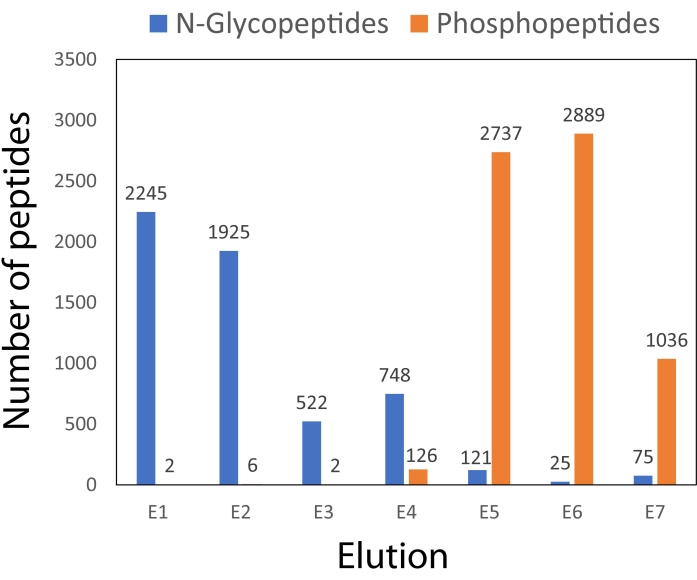
Figure 3: Comparison of peptide identifications from dual-functional Ti(IV)-IMAC enrichment across seven elutions. The number of identifications includes peptides identified in at least one of the two technical (injection) replicates. Please click here to view a larger version of this figure.
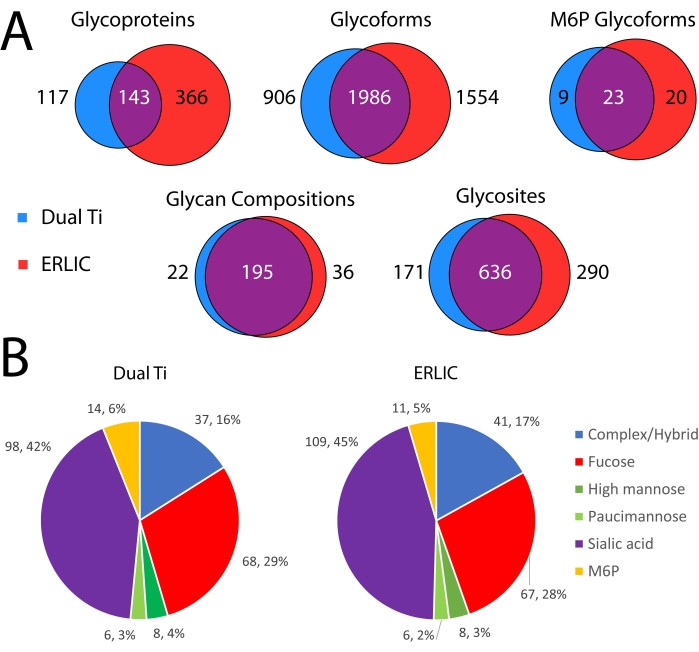
Figure 4: Comparison of glycoproteomics results from dual-functional Ti(IV)-IMAC enrichment compared to a conventional ERLIC glycopeptide enrichment. (A) Venn diagrams of glycoprotein, glycoform (protein, site, glycan), glycan composition, and glycosite identifications between enrichment methods. (B) Pie charts of glycans identified on peptide backbones between enrichment methods. Glycans are binned into six groups based on their composition16. Please click here to view a larger version of this figure.
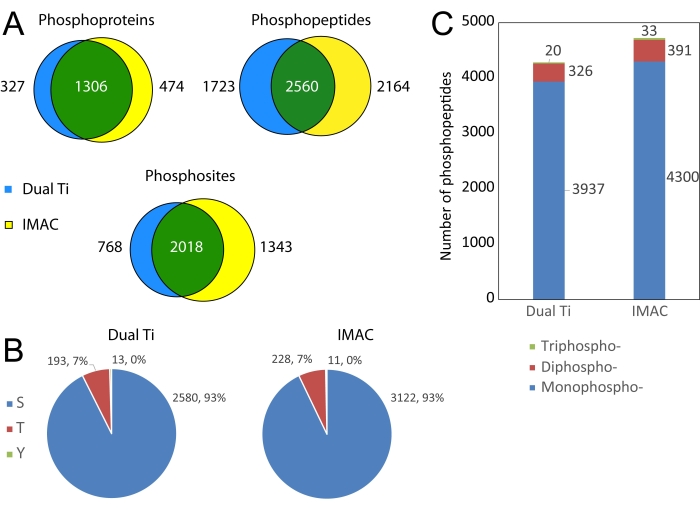
Figure 5: Comparison of phosphoproteomics results from dual-functional Ti(IV)-IMAC enrichment compared to a conventional Ti(IV)-IMAC phosphopeptide enrichment. (A) Venn diagrams of phosphoprotein, phosphopeptide, and phosphosite identifications between enrichment methods. (B) Pie charts of confidently localized (75% or higher probability) phosphosites broken down by amino acid residue. (C) Stacked bar plot of identified phosphopeptides binned by a number of phosphorylated residues. Please click here to view a larger version of this figure.
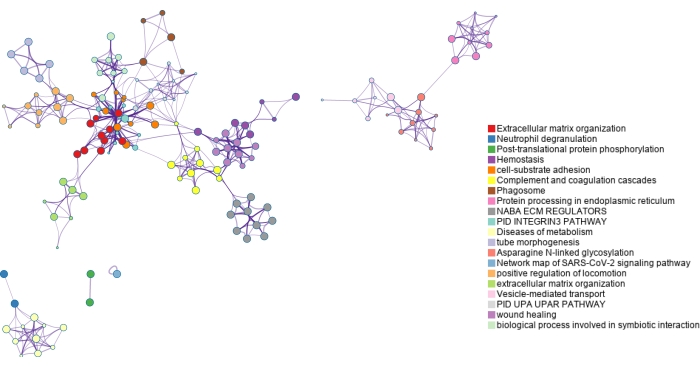
Figure 6: Gene ontology enrichment of N-glycoproteins identified using dual-functional Ti(IV)-IMAC enrichment. N-glycoproteins are mapped back to genes and significantly enriched pathways and process terms are identified using the entirety of the genome as the background. Various colors are used to label clusters of terms which are connected by term similarity. Please click here to view a larger version of this figure.
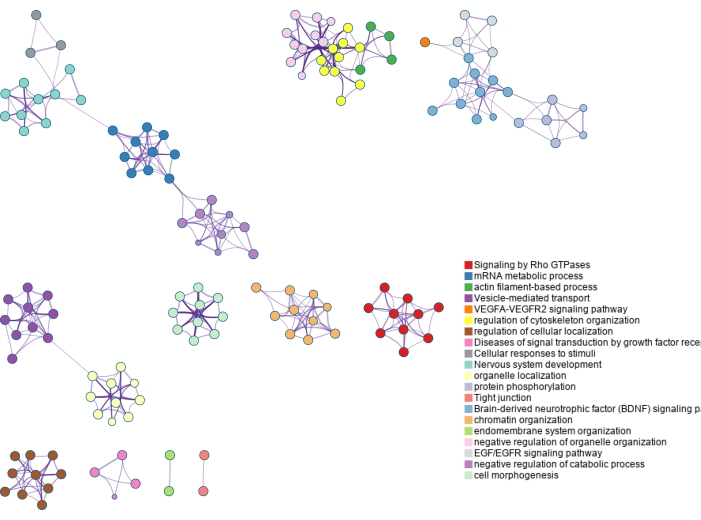
Figure 7: Gene ontology enrichment of phosphoproteins identified using dual-functional Ti(IV)-IMAC enrichment. Phosphoproteins are mapped back to genes and significantly enriched pathways and process terms are identified using the entirety of the genome as the background. Various colors are used to label clusters of terms, which are connected by term similarity. Please click here to view a larger version of this figure.
Supplementary Figure S1: Example total ion chromatograms (TICs) from each elution from dual-functional Ti(IV)-IMAC (E1 through E7, panels A-G) enrichment. Total signal (ion current) is plotted over the 117 min data collection period. Intensity of the base peak is given by the value NL. Please click here to download this File.
Supplementary Figure S2: Example total ion chromatograms (TICs) from each elution from ERLIC (E1 through E5, panels A-E) enrichment. Total signal (ion current) is plotted over the 117 min data collection period. Intensity of the base peak is given by the value NL. Please click here to download this File.
Supplementary Figure S3: Example total ion chromatogram (TIC) from the elution from Ti-IMAC enrichment. Total signal (ion current) is plotted over the 117 min data collection period. Intensity of the base peak is given by the value NL. Please click here to download this File.
Supplementary Figure S4: Gene ontology enrichment of N-glycoproteins identified using conventional glycopeptide enrichment with ERLIC. N-glycoproteins are mapped back to genes and significantly enriched pathways and process terms are identified using the entirety of the genome as the background. Various colors are used to label clusters of terms, which are connected by term similarity. Please click here to download this File.
Supplementary Figure S5: Gene ontology enrichment of phosphoproteins identified using conventional phosphopeptide enrichment with Ti(IV)-IMAC. Phosphoproteins are mapped back to genes and significantly enriched pathways and process terms are identified using the entirety of the genome as the background. Various colors are used to label clusters of terms, which are connected by term similarity. Please click here to download this File.
Supplementary Tables S1–S4: Metascape node information for gene ontology enrichment analyses. Tables contain node information for lists of N-glycoproteins and phosphoproteins identified using dual Ti (Table S1 and Table S2), N-glycoproteins using ERLIC (Table S3), and phosphoproteins using IMAC (Table S4). Please click here to download this Table.
