Here, a detailed method for the isolation and purification of amyloid fibrils using a modified sucrose density gradient ultracentrifugation purification method is summarized (see Figure 1). The innovation in this method is the inclusion of steps of ultrasonication-based washing using a water bath sonication system followed by SDS solubilization, which removes many loosely associated proteins from the amyloid fibrils that co-purify with the highly dense and clean fibrils. The ultrasonication step generates a high shearing force and agitates the fibrils, loosening the hydrophobic forces and releasing the SDS soluble loosely associated proteins into the SDS wash buffer. In turn, small quantities of highly pure amyloid fibril cores are recovered. As shown in Figure 2A, a visible pellet, which initially appears opaque (possibly due to impurities), can be seen after enrichment; however, following the ultrasonication and multiple SDS washes, the pellet turns semi-transparent and is hardly visible. The representative Congo red staining of purified amyloids as compared to the SDS soluble fraction documents the enrichment of the amyloid fibrils (Figure 2B). Congo red staining can be used to confirm the amyloid material in different fractions and can be visualized using a bright field microscope. As shown in Figure 2C, the SDS soluble fraction does not stain with the Congo red dye.
The structure of the purified fibrils with negative staining transmission electron microscopy analysis confirmed the presence of nearly pure amyloid fibrils (Figure 2D). Additionally, immunogold labeling using a combination of Aβ42 (6E10 and 4G8) antibodies confirmed the presence of Aβ42 peptides (Figure 2E). To investigate the composition and structural features of the purified material, we used immunoblot techniques for Aβ peptides and hallmark structural signatures (e.g., fibrils). Representative dot blot analysis of the fractions collected during amyloid isolation showed a relative abundance of Aβ42 peptides and fibrils using anti-Aβ42 and anti-fibril (LOC) antibodies (Figure 3A). Similarly, the western blot of the representative fractions also showed enrichment of Aβ42-containing fibrils in high molecular weight proteins trapped in the wells of the SDS PAGE gel (Figure 3B). To understand the composition of these high molecular weight fibrils, purified amyloid fractions were subjected to MS-based proteomic analysis. These semi-quantitative results revealed the presence of approximately 250 proteins, while the fraction collected before ultrasonication and SDS washes contained more than 2500 proteins (Figure 3C). This indicates the effectiveness of these two crucial steps that are included in this purification protocol. Taken together, multiple independent results indicate the high abundance of similar protein classes in fibril cores.
Gene Ontology (GO) cellular component analysis for one representative MS dataset in Figure 4 revealed that a large number of proteins present in the fibril cores are associated with non-membrane-bound organelle and supramolecular complexes. This observation is likely due to the inherent tendency of many proteins to aggregate themselves or co-aggregate with other proteins in proteinaceous inclusions that are in close proximity. Physical forces play crucial roles in these interactions. The other cellular organelle and components primarily represented by these proteins are mitochondria, cytoskeleton, cell membrane, and myelin sheath. Many of these proteins interact with Aβ peptides, oligomers, or protofibrils at different stages of amyloid formation. They might interact close to the plasma membrane, where Aβ peptides are released. The interaction may also happen while some of these proteins are released directly or via vesicular transport into the extracellular space. Secretion or exocytosis of protein aggregates is one among many strategies that cells use to cope and reduce the burden associated with protein aggregates15. This leaves another opportunity where some of the intracellular proteins can bind to Aβ peptides. The protocol provides a framework for further optimization depending on the experimental goals. For example, the purity and yield, by altering the number of ultrasonication and SDS washes, can be fine-tuned accordingly.
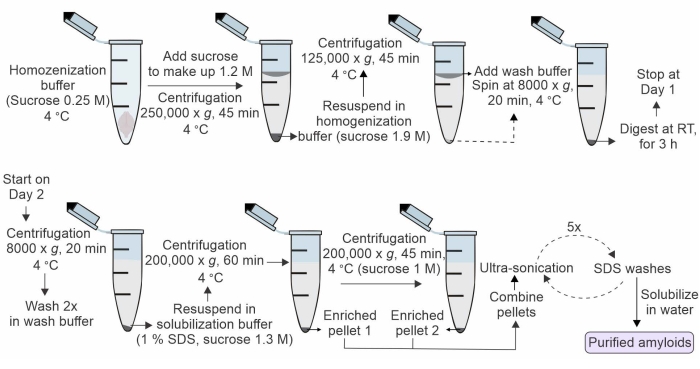
Figure 1: A diagrammatic overview of the workflow for isolation of amyloid fibrils core from AD post-mortem human or model animal brain tissues. Please click here to view a larger version of this figure.
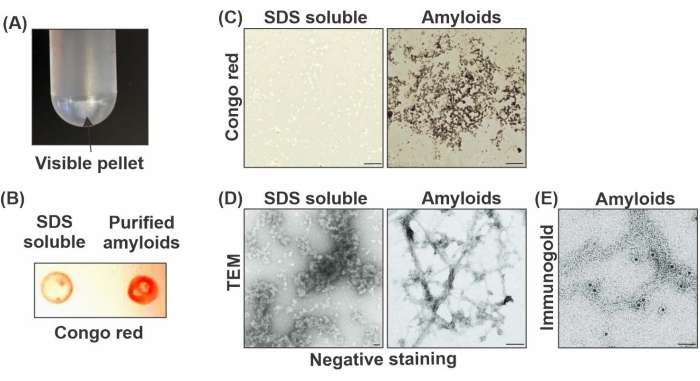
Figure 2: Confirmation of amyloid extraction using biochemical staining and imaging of amyloid fibrils. (A) Enriched amyloid-containing SDS insoluble pellet appears opaque off-white in color. (B) Congo red staining of SDS soluble supernatant and SDS insoluble pellet containing purified amyloid blotted on 0.45 µm nitrocellulose membrane; BCA readings were used for normalization of the loading amount of the proteins. BCA assay was performed as per the manufacturer's instructions. (C) Bright-field images of SDS soluble and amyloid material following Congo red staining (scale bar: 100 µm). (D) Visualization of SDS soluble fraction and purified amyloid fibrils using negative staining under the electron microscope (scale bar: 100 nm). (E) Confirmation of Aβ42 peptides abundance in purified amyloid fibrils by immunogold electron microscopy using Aβ42 (6E10 and 4G8) antibodies (scale bar: 50 nm). Please click here to view a larger version of this figure.
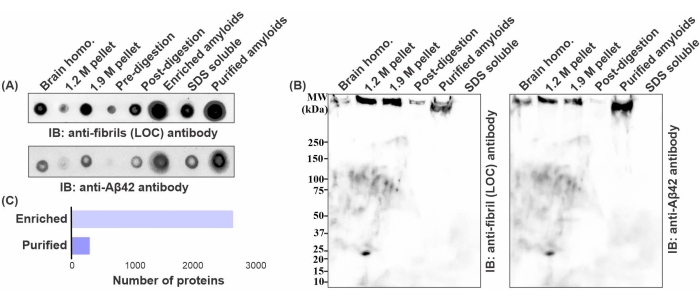
Figure 3: Validation of amyloid purification using immunoblot and MS analysis. (A) Dot blot and (B) Western blot analysis of several representative fractions collected during the amyloid purification process using anti-fibril LOC and anti-Aβ42 antibody; BCA assay readings were used for normalization of the loading amount of the proteins. (C) Number of proteins recovered in label-free mass spectrometry analysis of enriched and purified amyloid fractions; micro BCA assay was used for loading 3 µg of digested peptides for each MS analysis. BCA and micro BCA assays were performed as per the manufacturer's instructions. Please click here to view a larger version of this figure.
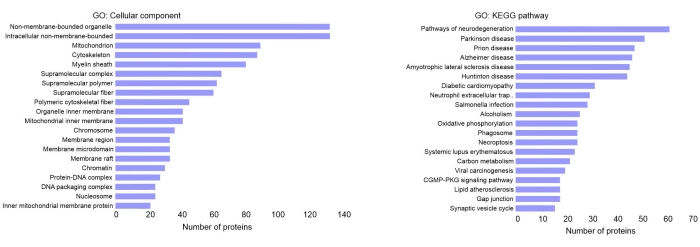
Figure 4: Gene Ontology analysis of proteins abundant in purified amyloid fractions. (A) Cellular components and (B) KEGG pathways. Please click here to view a larger version of this figure.
Supplementary File 1: Buffers and solutions, mass spectrometry parameters and ProLuCID search parameters for identification of peptides. Please click here to download this File.
Supplementary Table 1- A representative list of m/z ratios identified in MS run for amyloid beta peptides of APP knock in mouse models. Please click here to download this Table.
