In the electroporation setup, electrode positioning can play a role in the domain of transfection. The positive electrode is placed under the yolk, and the negative above the embryo (Figure 1A). This results in higher GFP expression in much of the inner ear and both vestibular organs (Figure 1B), and auditory basilar papilla (Figure 1C,D), confirming transfection.
In assessing the phenotype of CRISPR-knockdowns, we designed guide RNAs to the hair cell transcription factor Atonal homolog 1 (Atoh1). Mouse mutants of Atoh1 are unable to form hair cells32; after electroporation of Atoh1 guide RNAs and incubation until E10, we find that HC development is impaired when compared to control, empty plasmid control (Figure 2). Although electroporation is mosaic (Figure 2E,F), control electroporated cells are able to form hair cells. In Atoh1 gRNA electroporated samples, GFP positive cells never show the markers of HC development (Figure 2B).
Organ culture of the BP provides accessibility to the tissue. Cultures in a 3D matrix, such as collagen, provide excellent preservation of tissue morphology for up to 5 days. The organization of HC and SC is maintained in these culture conditions (Figure 3).
Organ cultures on a membrane are preferable for imaging stereocilia. Such cultures can be cultured for up to 5 days while maintaining the hair bundle integrity. This can be seen by the localization of the tip-link protein, protocadherin 1533 (Pcdh15) (Figure 4). For interrogating the development of the hair bundle, higher resolution imaging is necessary, and such approaches, using either super-resolution microscopy (Figure 4D) or scanning electron microscopy (Figure 4E), provide more complete information.
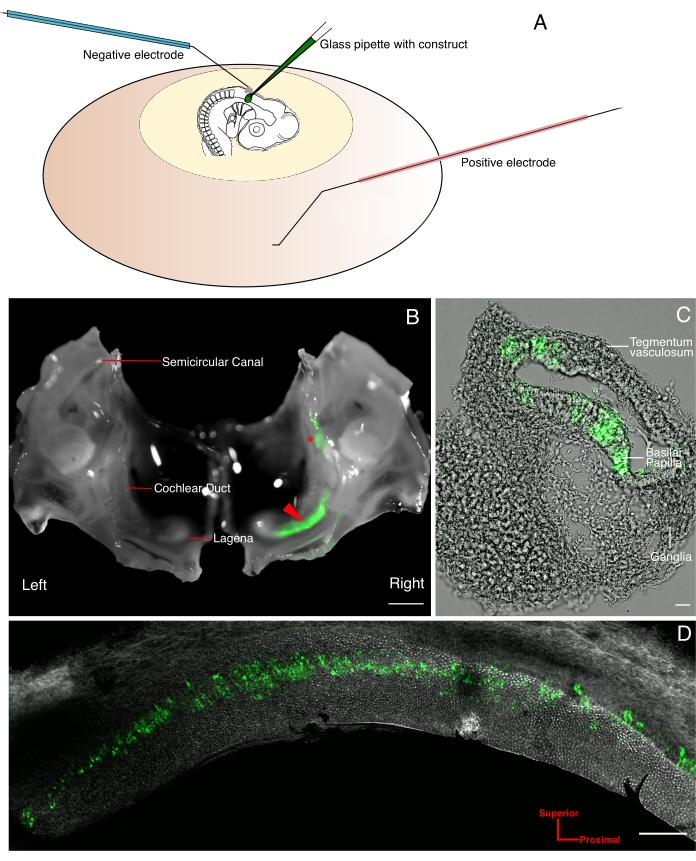
Figure 1. GFP expression is visible at E10 after in ovo electroporation at E4. (A) Schematic diagram illustrating in ovo microinjection and electroporation in chick otic vesicle at E4. The injection pipette is filled with Tol2-eGFP (T2K-eGFP) and Tol2-transposases (T2TP) plasmids, together with fast green dye for visualization. (B) Image of the electroporated inner ear at E10 using a 0.63x air objective lens on a stereomicroscope. The left inner ear is the internal control. The red arrow marks the GFP expression in the cochlear duct of the right inner ear, and the red asterisk shows GFP expression in vestibular organs; scale bar is 2 cm. (C) Widefield fluorescence image of a cross-section of right cochlear duct using a 20x air objective of 0.5 NA. GFP expression is mostly confined to the sensory epithelium; scale bar is 10 µm. (D) Confocal image of whole mount basilar papilla imaged using 10x air objective of 0.5 NA stained with phalloidin conjugated with Alexa 647 fluorophore. GFP expression is observed from proximal to distal end on the neural side of the BP; scale bar is 100 µm. Please click here to view a larger version of this figure.
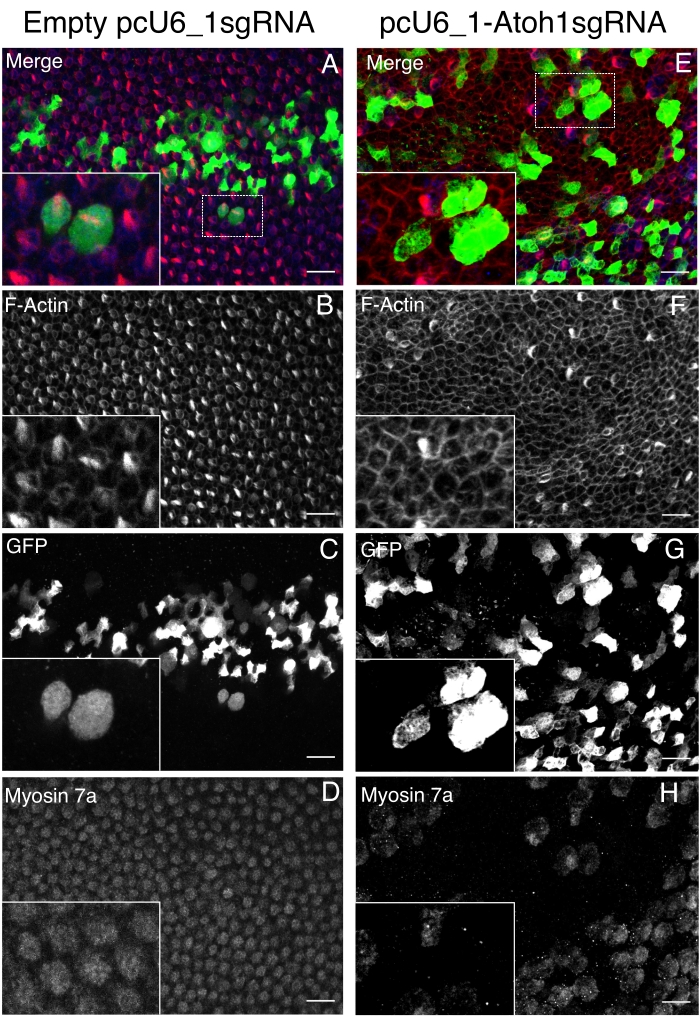
Figure 2. CRISPR/Cas9-mediated Atoh1 gene knockout viain ovo electroporation results in loss of hair cells (HCs). Atoh1 gene guide plasmid pcU6_1-Atoh1sgRNA and tracer plasmids Tol2-eGFP (T2K-eGFP) and Tol2-transposases (T2TP) with SpCas9 protein are microinjected and electroporated in chick otic vesicle at E4. All the images are from chick E10 basilar papilla with a 10 µm scale bar, captured with a 60x oil immersion objective of 1.42 NA using a laser confocal microscope. Zoomed in insets are provided for all images. (A,B,C,D) The left-hand panel contains BP with hair cells (HCs), electroporated with empty pcU6_1sgRNA and T2K-eGFP, and T2TP with SpCas9 protein. (E,F,G,H) The right-hand side panel contains BP with loss of HCs, electroporated with Atoh1 guide (pcU6_1-Atoh1sgRNA) and T2K-eGFP, and T2TP with SpCas9 protein. (A,E) Merged image showing hair cells (HCs) in the basilar papilla. These are immunoreactive for Myosin 7a (blue); F-actin stained with phalloidin conjugated with Alexa 647 (red); GFP expression from Tol2-eGFP plasmids is detected using an anti-GFP antibody (green). The loss of HCs is evident from Myosin 7a immunoreactivity when comparing treatments with empty pcU6_1sgRNA (D) and pcU6_1-Atoh1sgRNA (H). F-actin imaging from both treatments highlights the hair bundle of the HC (B,F). (C,G) T2K-eGFP and T2TP are used to measure transfection location and efficiency. Please click here to view a larger version of this figure.
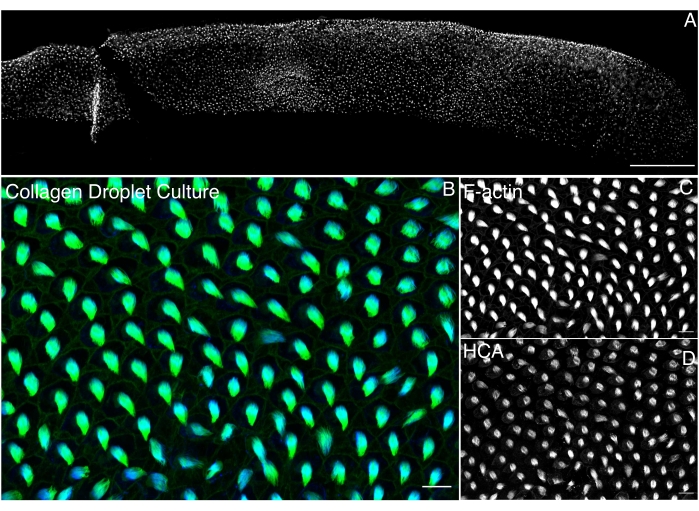
Figure 3. Organ culture of cochlear duct in 3D-collagen droplet culture maintains the organization of sensory epithelium. The BP at E10 is cultured in a 3D collagen droplet culture for 1 day and imaged using 60x oil immersion objective of 1.42 NA using a laser confocal microscope. (A) Image of whole BP of E10 cultured for 1 day in a collagen droplet and stained with antibodies against hair cell antigen (HCA)34. Scale bar is 100 µm. (B) Merged image showing the preserved organization of sensory epithelium from the distal side of BP stained with (C) phalloidin (green) and (D) HCA (blue); scale bar is 10 µm. Please click here to view a larger version of this figure.
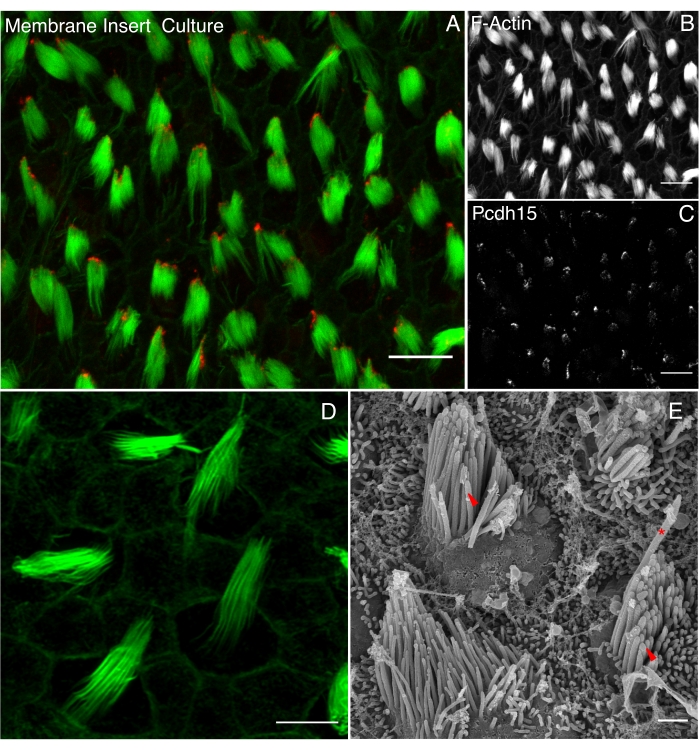
Figure 4. Stereociliary bundles of basilar papilla under electron and light microscope. 0.1% dimethylsulphoxide (DMSO)-treated BP were explanted at E10 and cultured for 3 days in vitro (DIV) on membrane culture inserts. (A) Explant is imaged using a 60x oil immersion objective of 1.42 NA with a laser scanning confocal microscope. The merged image shows the expression of the protocadherin 15 (Pcdh15) and stereocilia marked by phalloidin conjugated with Alexa 488 (green). Single-channel images of F-actin (B) and Pcdh15 (C) are shown. Scale bar is 10 µm. (D) Super-resolution image of stereocilia stained with phalloidin conjugated with Alexa 488 staining in green. The image was obtained using a 63x oil immersion objective of 1.42 NA in Airyscan mode of laser scanning confocal microscope. Scale bar is 5 µm. (E) SEM image was taken at 16340x magnification using 7 kV electron high tension (EHT) voltage. Red asterisk marks kinocilia and red wedge marks stereocilia. Brightness and contrast were adjusted to auto and the image was sharpened using FIJI. Scale bar is 1 µm. Please click here to view a larger version of this figure.
