تم استخدام RNA-seq على نطاق واسع على مر السنين عادة لتقدير التعبير الجيني التفاضلي واكتشاف الجينات1. بالإضافة إلى ذلك ، يمكن استخدامه أيضا لتقدير الاستخدام المتغير لمستوى إكسون بسبب تعبير الجينات عن أشكال متساوية مختلفة ، وبالتالي المساهمة في فهم أفضل لتنظيم الجينات على مستوى ما بعد النسخ. تولد غالبية الجينات حقيقية النواة أشكالا متساوية مختلفة عن طريق الربط البديل (AS) لزيادة تنوع تعبير mRNA. يمكن تقسيم أحداث AS إلى أنماط مختلفة: تخطي exons كاملة (SE) حيث تتم إزالة exon (“كاسيت”) تماما من النص جنبا إلى جنب مع الإنترونات المرافقة له ؛ اختيار موقع لصق بديل (مانح) 5 ‘(A5SS) واختيار موقع لصق بديل 3 ‘(متقبل) (A3SS) عند وجود موقعين أو أكثر من مواقع لصق على طرفي إكسون ؛ الاحتفاظ بالإنترونات (RI) عند الاحتفاظ ب intron ضمن نسخة mRNA الناضجة والاستبعاد المتبادل لاستخدام exon (MXE) حيث يمكن الاحتفاظ بواحد فقط من الإكسونات المتاحين في وقت 2,3. يلعب polyadenylation البديل (APA) أيضا دورا مهما في تنظيم التعبير الجيني باستخدام مواقع poly البديلة (A) لتوليد أشكال متماثلة متعددة من mRNA من نسخة واحدة4. تقع معظم مواقع polyadenylation (pAs) في المنطقة غير المترجمة 3 ‘(3’ UTRs) ، مما يولد أشكالا متماثلة من mRNA بأطوال UTR متنوعة 3 بوصات. نظرا لأن 3 ‘UTR هو المحور المركزي للتعرف على العناصر التنظيمية ، يمكن أن تؤثر أطوال UTR المختلفة 3 ‘على توطين mRNA واستقراره وترجمته5. هناك فئة من 3 ‘مقايسات تسلسل نهاية محسنة للكشف عن APA التي تختلف في تفاصيل البروتوكول6. تم تصميم خط الأنابيب الموصوف هنا ل PolyA-seq ، ولكن يمكن تكييفه مع البروتوكولات الأخرى كما هو موضح.
في هذه الدراسة ، نقدم مجموعة من طرق تحليل exon التفاضلية 7,8 (الشكل 1) ، والتي يمكن تقسيمها إلى فئتين عريضتين: القائمة على exon (DEXSeq9 ، diffSplice 10) والقائمة على الحدث (تكرار التحليل متعدد المتغيرات لربط النص (rMATS)11). تقارن الطرق القائمة على الإكسون تغير الطي عبر ظروف الإكسونات الفردية ، مقابل مقياس للتغير الكلي في طية الجينات لاستدعاء استخدام إكسون المعبر عنه بشكل تفاضلي ، ومن ذلك تحسب مقياسا على مستوى الجينات لنشاط AS. تستخدم الطرق المستندة إلى الأحداث قراءات تقاطع exon-intron-spanning لاكتشاف وتصنيف أحداث الربط المحددة مثل تخطي exon أو الاحتفاظ بالإنترونات ، وتمييز أنواع AS هذه في الإخراج3. وبالتالي ، توفر هذه الطرق وجهات نظر تكميلية لتحليل كامل لمعيار المحاسبة12,13. اخترنا DEXSeq (استنادا إلى حزمة DESeq214 DGE) و diffSplice (استنادا إلى حزمة Limma10 DGE) للدراسة لأنهما من بين الحزم الأكثر استخداما لتحليل الربط التفاضلي. تم اختيار rMATS كطريقة شائعة للتحليل القائم على الأحداث. طريقة أخرى شائعة قائمة على الأحداث هي MISO (خليط من الأشكال المتساوية)1. بالنسبة إلى APA ، نقوم بتكييف النهج القائم على exon.
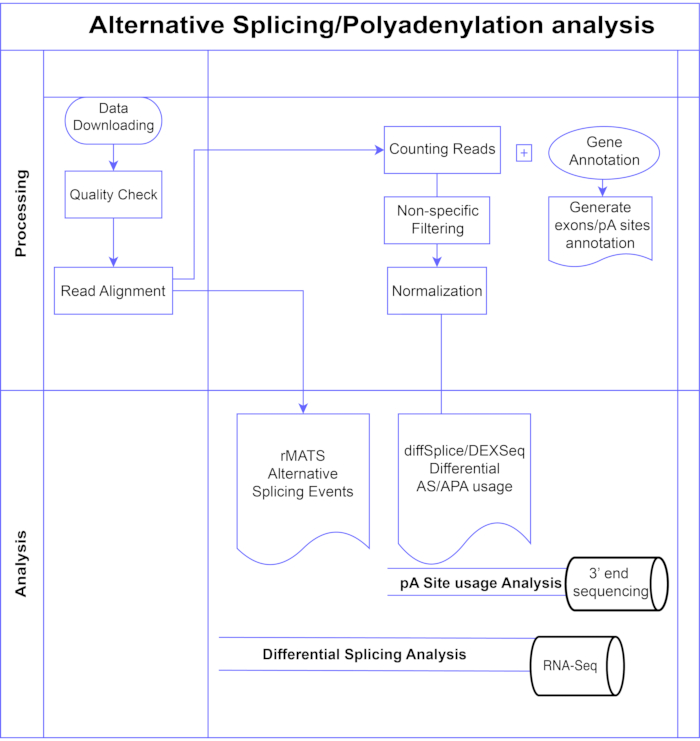
الشكل 1. خط أنابيب التحليل. مخطط انسيابي للخطوات المستخدمة في التحليل. تشمل الخطوات: الحصول على البيانات ، وإجراء فحوصات الجودة ومحاذاة القراءة متبوعة بحساب القراءات باستخدام التعليقات التوضيحية لمواقع exons و introns و pA المعروفة ، والتصفية لإزالة الأعداد المنخفضة والتطبيع. تم تحليل بيانات PolyA-seq لمواقع pA البديلة باستخدام طرق diffSplice / DEXSeq ، وتم تحليل RNA-Seq السائب للربط البديل على مستوى exon باستخدام طرق diffSplice / DEXseq ، وتم تحليل أحداث AS باستخدام rMATS. الرجاء الضغط هنا لعرض نسخة أكبر من هذا الشكل.
تم الحصول على بيانات RNA-seq المستخدمة في هذا المسح من التعبير الجيني الجامع (GEO) (GSE138691)15. استخدمنا بيانات RNA-seq للفأر من هذه الدراسة مع مجموعتين من الحالات: النوع البري (WT) والضربة القاضية من النوع 1 الشبيه بالعضلات (Mbnl1 KO) مع ثلاث نسخ متماثلة لكل منهما. لإثبات تحليل استخدام موقع polyadenylation التفاضلي ، حصلنا على بيانات الخلايا الليفية الجنينية للفأر (MEFs) PolyA-seq (GEO Accession GSE60487)16. تحتوي البيانات على أربع مجموعات شروط: النوع البري (WT) ، النوع 1 / النوع 2 الشبيه بالعضلات بالضربة القاضية المزدوجة (Mbnl1/2 DKO) ، Mbnl 1/2 DKO مع ضربة قاضية Mbnl3 (KD) و Mbnl1/2 DKO مع التحكم Mbnl3 (Ctrl). تتكون كل مجموعة شرط من نسختين متماثلتين.
| انضمام GEO | رقم تشغيل SRA | اسم العينة | شرط | تكرار | نسيج | التسلسل | طول القراءة | |
| RNA-Seq | جي إس إم 4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 بالضربة القاضية | ممثل 1 | الغدة الصعترية | نهاية مزدوجة | 100 نقطة أساس |
| جي إس إم 4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 بالضربة القاضية | ممثل 2 | الغدة الصعترية | نهاية مزدوجة | 100 نقطة أساس | |
| جي إس إم 4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 بالضربة القاضية | مندوب 3 | الغدة الصعترية | نهاية مزدوجة | 100 نقطة أساس | |
| جي إس إم 4116221 | SRR10261604 | WT_Thymus_1 | نوع البرية | ممثل 1 | الغدة الصعترية | نهاية مزدوجة | 100 نقطة أساس | |
| جي إس إم 4116222 | SRR10261605 | WT_Thymus_2 | نوع البرية | ممثل 2 | الغدة الصعترية | نهاية مزدوجة | 100 نقطة أساس | |
| جي إس إم 4116223 | SRR10261606 | WT_Thymus_3 | نوع البرية | مندوب 3 | الغدة الصعترية | نهاية مزدوجة | 100 نقطة أساس | |
| 3P-Seq | جي إس إم 1480973 | ريال1553129 | WT_1 | النوع البري (WT) | ممثل 1 | الخلايا الليفية الجنينية للفأر (MEFs) | نهاية واحدة | 40 نقطة أساس |
| جي إس إم 1480974 | ريال1553130 | WT_2 | النوع البري (WT) | ممثل 2 | الخلايا الليفية الجنينية للفأر (MEFs) | نهاية واحدة | 40 نقطة أساس | |
| جي إس إم 1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 ضربة قاضية مزدوجة (DKO) | ممثل 1 | الخلايا الليفية الجنينية للفأر (MEFs) | نهاية واحدة | 40 نقطة أساس | |
| جي إس إم 1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 ضربة قاضية مزدوجة (DKO) | ممثل 2 | الخلايا الليفية الجنينية للفأر (MEFs) | نهاية واحدة | 40 نقطة أساس | |
| جي إس إم 1480977 | ريال1553133 | DKOsiRNA_1 | Mbnl 1/2 ضربة قاضية مزدوجة مع Mbnl 3 siRNA (دينار كويتي) | ممثل 1 | الخلايا الليفية الجنينية للفأر (MEFs) | نهاية واحدة | 40 نقطة أساس | |
| جي إس إم 1480978 | ريال1553134 | DKOsiRNA_2 | Mbnl 1/2 ضربة قاضية مزدوجة مع Mbnl 3 siRNA (دينار كويتي) | ممثل 2 | الخلايا الليفية الجنينية للفأر (MEFs) | نهاية واحدة | 36 نقطة أساس | |
| جي إس إم 1480979 | ريال1553135 | DKONTsiRNA_1 | Mbnl 1/2 ضربة قاضية مزدوجة مع siRNA غير مستهدف (Ctrl) | ممثل 1 | الخلايا الليفية الجنينية للفأر (MEFs) | نهاية واحدة | 40 نقطة أساس | |
| جي إس إم 1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 ضربة قاضية مزدوجة مع siRNA غير مستهدف (Ctrl) | ممثل 2 | الخلايا الليفية الجنينية للفأر (MEFs) | نهاية واحدة | 40 نقطة أساس |
الجدول 1. ملخص مجموعات بيانات RNA-Seq و PolyA-seq المستخدمة في التحليل.
