L’RNA-seq è stato ampiamente utilizzato nel corso degli anni, tipicamente per stimare l’espressione genica differenziale e la scopertagenica 1. Inoltre, può anche essere utilizzato per stimare l’uso variabile del livello di esone a causa del gene che esprime diverse isoforme, contribuendo così a una migliore comprensione della regolazione genica a livello post-trascrizionale. La maggior parte dei geni eucariotici genera diverse isoforme mediante splicing alternativo (AS) per aumentare la diversità dell’espressione dell’mRNA. Gli eventi AS possono essere suddivisi in diversi modelli: salto di esoni completi (SE) in cui un esone (“cassetta”) viene completamente rimosso dalla trascrizione insieme ai suoi introni laterali; selezione alternativa (donatore) del sito di giunzione 5′ (A5SS) e selezione alternativa del sito di giunzione 3′ (accettore) (A3SS) quando due o più siti di giunzione sono presenti su entrambe le estremità di un esone; ritenzione degli introni (RI) quando un introne viene trattenuto all’interno del trascritto dell’mRNA maturo e mutua esclusione dell’uso dell’esone (MXE) in cui solo uno dei due esoni disponibili può essere trattenuto alla volta 2,3. La poliadenilazione alternativa (APA) svolge anche un ruolo importante nella regolazione dell’espressione genica utilizzando siti di poli alternativi (A) per generare più isoforme di mRNA da una singola trascrizione4. La maggior parte dei siti di poliadenilazione (pAs) si trovano nella regione 3′ non tradotta (3′ UTR), generando isoforme di mRNA con diverse lunghezze UTR 3′. Poiché l’UTR 3′ è l’hub centrale per il riconoscimento degli elementi regolatori, diverse lunghezze UTR 3′ possono influenzare la localizzazione, la stabilità e la traduzione dell’mRNA5. Esistono saggi di sequenziamento finale di classe 3′ ottimizzati per rilevare APA che differiscono nei dettagli del protocollo6. La pipeline qui descritta è progettata per PolyA-seq, ma può essere adattata per altri protocolli come descritto.
In questo studio, presentiamo una pipeline di metodi di analisi differenziale degli esoni7,8 (Figura 1), che possono essere suddivisi in due grandi categorie: basati sull’esone (DEXSeq9, diffSplice 10) e basati sugli eventi (replicate Multivariate Analysis of Transcript Splicing (rMATS)11). I metodi basati sull’esone confrontano il cambiamento di piega tra le condizioni dei singoli esoni, contro una misura del cambiamento complessivo della piega genica per chiamare l’uso differenziale dell’esone, e da ciò calcolare una misura a livello genetico dell’attività AS. I metodi basati su eventi utilizzano letture di giunzione esone-introne per rilevare e classificare eventi di splicing specifici come il salto dell’esone o la ritenzione di introni e distinguere questi tipi di AS nell’output3. Pertanto, questi metodi forniscono punti di vista complementari per un’analisi completa di AS12,13. Abbiamo selezionato DEXSeq (basato sul pacchetto DESeq214 DGE) e diffSplice (basato sul pacchetto Limma10 DGE) per lo studio in quanto sono tra i pacchetti più utilizzati per l’analisi di splicing differenziale. rMATS è stato scelto come metodo popolare per l’analisi basata sugli eventi. Un altro metodo popolare basato su eventi è MISO (Mixture of Isoforms)1. Per l’APA adattiamo l’approccio basato sull’esone.
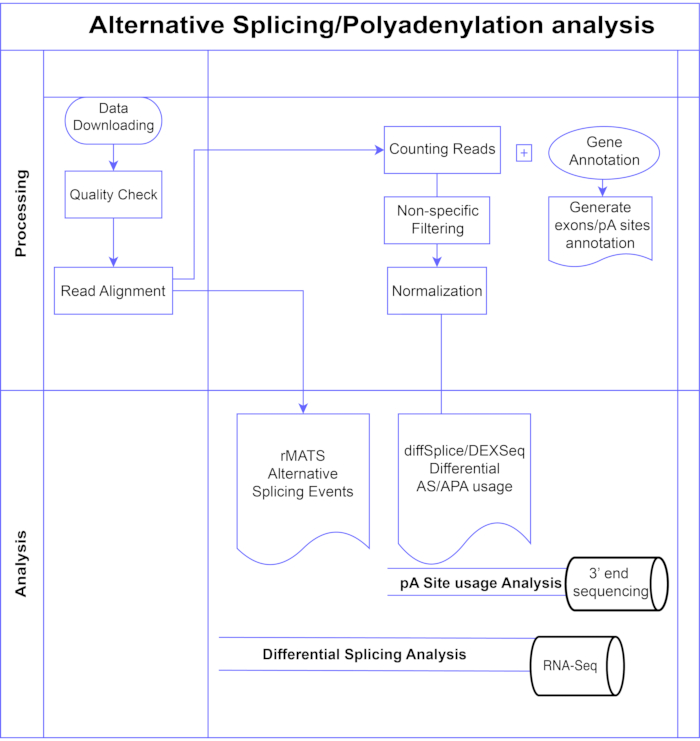
Figura 1. Pipeline di analisi. Diagramma di flusso dei passaggi utilizzati nell’analisi. I passaggi includono: ottenere i dati, eseguire controlli di qualità e allineamento delle letture seguito dal conteggio delle letture utilizzando annotazioni per esoni, introni e siti pA noti, filtraggio per rimuovere conteggi bassi e normalizzazione. I dati di PolyA-seq sono stati analizzati per siti pA alternativi utilizzando metodi diffSplice/DEXSeq, RNA-Seq di massa sono stati analizzati per lo splicing alternativo a livello di esone con metodi diffSplice/DEXseq e gli eventi AS analizzati con rMATS. Fare clic qui per visualizzare una versione ingrandita di questa figura.
I dati RNA-seq utilizzati in questa indagine sono stati acquisiti da Gene Expression Omnibus (GEO) (GSE138691)15. Abbiamo utilizzato i dati RNA-seq di topo di questo studio con due gruppi di condizioni: wild-type (WT) e Muscleblind-like type 1 knockout (Mbnl1 KO) con tre repliche ciascuno. Per dimostrare l’analisi differenziale dell’utilizzo del sito di poliadenilazione, abbiamo ottenuto dati PolyA-seq di fibroblasti embrionali di topo (MEF) (GEO Accession GSE60487)16. I dati hanno quattro gruppi di condizioni: Wild-type (WT), Muscleblind-like type1/type 2 double knockout (Mbnl1/2 DKO), Mbnl 1/2 DKO con Mbnl3 knockdown (KD) e Mbnl1/2 DKO con controllo Mbnl3 (Ctrl). Ogni gruppo di condizioni è costituito da due repliche.
| Adesione GEO | Numero di esecuzione SRA | Nome del campione | Condizione | Replicare | Fazzoletto | Sequenziamento | Lunghezza di lettura | |
| RNA-Seq | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 knockout | Rappresentante 1 | Timo | Estremità accoppiata | 100 pb |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 knockout | Rappresentante 2 | Timo | Estremità accoppiata | 100 pb | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 knockout | Rappresentante 3 | Timo | Estremità accoppiata | 100 pb | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | Tipo selvaggio | Rappresentante 1 | Timo | Estremità accoppiata | 100 pb | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | Tipo selvaggio | Rappresentante 2 | Timo | Estremità accoppiata | 100 pb | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | Tipo selvaggio | Rappresentante 3 | Timo | Estremità accoppiata | 100 pb | |
| 3P-Seq | GSM1480973 | SRR1553129 | WT_1 | Tipo selvatico (WT) | Rappresentante 1 | Fibroblasti embrionali di topo (MEF) | Estremità singola | 40 pb |
| GSM1480974 | SRR1553130 | WT_2 | Tipo selvatico (WT) | Rappresentante 2 | Fibroblasti embrionali di topo (MEF) | Estremità singola | 40 pb | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 doppio knockout (DKO) | Rappresentante 1 | Fibroblasti embrionali di topo (MEF) | Estremità singola | 40 pb | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 doppio knockout (DKO) | Rappresentante 2 | Fibroblasti embrionali di topo (MEF) | Estremità singola | 40 pb | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 doppio knockout con Mbnl 3 siRNA (KD) | Rappresentante 1 | Fibroblasti embrionali di topo (MEF) | Estremità singola | 40 pb | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 doppio knockout con Mbnl 3 siRNA (KD) | Rappresentante 2 | Fibroblasti embrionali di topo (MEF) | Estremità singola | 36 pb | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 doppio knockout con siRNA non mirato (Ctrl) | Rappresentante 1 | Fibroblasti embrionali di topo (MEF) | Estremità singola | 40 pb | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 doppio knockout con siRNA non mirato (Ctrl) | Rappresentante 2 | Fibroblasti embrionali di topo (MEF) | Estremità singola | 40 pb |
Tabella 1. Riepilogo dei set di dati RNA-Seq e PolyA-seq utilizzati per l’analisi.
