Small-angle neutron scattering (SANS) provides a unique way to measure the sizes, shapes, interactions, and organization of various materials on length scales from ≈1 nm to ≈100 nm1,2,3. Recent instruments, including VSANS (very small-angle neutron scattering) instruments with focusing mirrors, push the limits toward measuring even larger length scales up to ≈1000 nm4,5. In general, the unique scattering contrast inherent to neutron scattering methods offers several advantages in measuring the time-evolution of nanoscale structures, such as the aggregation of components in pharmaceutical formulations6, crosslinking and gelation reactions in polymer systems7,8, in meso crystallization of membrane proteins9,10, degradation and unfolding of proteins11,12, and growth of silica-based materials13,14,15. The unique scattering contrast makes time-resolved SANS (TR-SANS) a useful complement to other stopped-flow-based measurements.
Stopped-flow mixing methods often are implemented in small-angle X-ray scattering (SAXS)16,17,18,19,20,21, fluorescence spectroscopy22,23,24,25,26, and light scattering27,28,29,30,31,32 experiments to study kinetic processes on the millisecond time scales. An important difference between SANS and SAXS is that neutron scattering is a nondestructive characterization technique, and as such, SANS can be used to measure the same sample for hours or even days without ionizing radiation damage to the sample, which can happen during higher-flux X-ray scattering experiments33. As repeated SANS measurements will not alter the chemical structure of the probe molecule or sample, the time-evolution can be studied without effects of photobleaching, for example, which can complicate kinetics measurements that rely on fluorescence23,24. Moreover, SANS can be used to measure highly concentrated and optically opaque samples that are often difficult to characterize with light-based techniques such as dynamic light scattering.
In addition to providing structural information on the nanoscale, SANS can be used to probe the local composition of these structures through the variation in neutron scattering length density contrast. The scattering length density (SLD) of different elements varies randomly across the periodic table and varies with different isotopes of the same element. A commonly exploited example is hydrogen (1H or H) and deuterium (2H or D), which have vastly different neutron scattering lengths. Therefore, hydrogen-rich materials, such as surfactants, lipids, proteins, RNA, DNA, and other polymers, can be distinguished from deuterated solvents using SANS without significantly changing the physical properties of the system. However, it is important to note that H/D exchange can affect the density, hydrogen-bonding, and phase transition temperatures in the sample. Nevertheless, the unique sensitivity of SANS to hydrogen-rich materials is especially useful in soft matter research where the samples of interest have lower scattering contrast and signal in X-ray-based techniques such as SAXS. Isotopic substitution also makes SANS a powerful tool for studying molecular exchange kinetics in hydrogen-rich materials by simply mixing H-labeled and D-labeled molecules. Isotopic substitution is particularly useful in systems where bulky fluorescent dyes are larger than the surfactant or lipid molecules of interest and can influence the exchange kinetics34,35.
Time-resolved SANS measurements are advantageous because the measured intensity is a function of time, length scale, and SLD contrast. As such, TR-SANS experiments can be designed to probe the time-dependent changes in the spatial distributions and the compositions of the samples. These unique advantages of SANS have led to important insights into kinetic processes in many soft material systems such as surfactants36,37,38, emulsions39,40,41, lipids34,42,43,44,45,46,47,48,49,50, and polymers51,52,53,54,55,56,57,58,59,60,61,62. Most TR-SANS studies have focused on time scales of minutes to hours. However, many kinetic processes of interest occur on the second time scale and are essential for understanding the underlying mechanisms. Capturing these early time points requires that the solutions be rapidly mixed and measured in situ, in which the mixing is synced with data collection during stopped-flow light scattering27,28,29,30,31,32, fluorescence22,23,24,25,26, and X-ray16,17,18,19,20,21 experiments. This work describes the use of a sample environment designed to rapidly mix multiple liquid samples and inject the mixture into a quartz glass cell for TR-SANS measurements. The mixing device is an adaptation of the recently developed capillary rheoSANS device63 and uses multiple syringe pumps and valves to control the sample mixing and to automate cell cleaning. By connecting syringe pumps to a series of flow selector valves, multiple inlet streams can be repeatedly mixed, measured, rinsed, and dried to facilitate TR-SANS measurements on the seconds time scale.
The current procedure assumes that the samples of interest have been identified and prepared. We focus on the in situ mixing setup and methods to collect TR-SANS data. Neutron scattering data were collected on the VSANS instrument at the NIST Center for Neutron Research (NCNR); however, the procedure should be applicable to other SANS instruments. Readers interested in implementing similar protocols on other SANS instruments should consult with the local instrument scientists to determine the optimal instrument configuration to maximize neutron flux at the desired length scale and time scale most relevant to the kinetic processes of interest. The data presented here were collected using the high flux 'white beam' configuration on VSANS to maximize neutron counts at the loss of spatial resolution5. The detector carriages were positioned to cover a range of scattering vectors (q), 0.005 Å-1 < q < 0.5 Å-1, corresponding to length scales of ≈130 nm to ≈13 nm. The scattering vector is defined as q = 4π/λ sin (θ/2) in which λ is the neutron wavelength, and θ is the scattering angle.
The stopped-flow mixing device used for the TR-SANS measurements consists of multiple pumps, rinsing syringes, sample syringes, flow selectors, as well as adynamic mixer, sample cell, and mixed sample container, as shown in Figure 1. All sealed fluid paths are located inside an air-conditioned enclosure, which includes the syringes, valves, connection tubing, dynamic mixer, and sample cells. A programmable thermoelectric air conditioner is used to control the enclosure temperature in the range from 10 °C to 50 °C within ±1 °C. Note that some of the enclosure insulation was removed to show the working parts of the device. The main mixing device enclosure is positioned on a translational stage on the NG3 VSANS beam line at the NCNR. The enclosure position is adjusted using the translation stage to position the sample cell in the path of the neutron beam (yellow dashed line).

Figure 1: An example setup for combining stopped-flow mixing and small-angle neutron scattering measurements at the VSANS beamline at the NIST Center for Neutron Research. The setup contains four syringe pumps, two syringes for solvent rinsing and two syringes for sample injection, four pump selector valves, two mixer selector valves, a dynamic mixer, a flow-through quartz cell, and a mixed sample container. Incident neutrons scatter off the mixed sample located inside the sample cell. An insulated enclosure with quartz windows and a thermoelectric air-conditioned unit is used to control the sample and all equipment at a constant temperature. The yellow dashed line shows the neutron beam path. Scale bar = 10 cm. Please click here to view a larger version of this figure.
The device depicted in Figure 1 is configured with two sample syringes, two rinsing syringes, and one sample cell. Corresponding flow diagrams for the different steps of the protocol are illustrated in Figure 2. The desired volumes of the two different samples are injected into the mixer and the sample cell (Figure 2A). Once the sample cell is filled, the Inlet Switch Valve (ISV) and Outlet Switch Valve (OSV) are closed to isolate the sample cell from the dynamic mixer and to prevent sample back diffusion into the cell during TR-SANS data collection (Figure 2B). Before the dynamic mixer, the connection tubing varies in length from 10 cm to 1 m and does not affect the mixing delay time. However, tubing connections between the dynamic mixer and the sample cell will affect the mixing delay time and the required sample injection volume. Precut stainless steel tubing with 0.04 inch (1 mm) inner diameter and 100 mm length are used to connect the dynamic mixer, the Mixer Selector Valves (MSV1 and MSV2), and the ISV and OSV. Fluorinated tubing with 1 mm inner diameter and 115 mm length is used to connect the ISV and OSV (or the dynamic mixer outlet) to the sample cell. The total void volume that influences the mixing delay time includes the mixer void volume (0.15 mL), the tubing between the mixer outlet and the sample cell inlet (0.09 mL), and the sample cell volume (0.16 mL). In this example, the total void volume is 0.4 mL. The internal void volumes of valves are negligible compared to the tubing, mixer, and sample cell void volumes. For example, the employed low-pressure selector valves (0.75 mm bore diameter) contain approximate void volumes of 4 µL, while the high-pressure selector valves and switch valves (0.25 mm bore diameter) contain approximate void volumes of 0.5 µL.
After the TR-SANS measurement is complete, the sample is pushed out of the cell with solvent, and rinse solvent is repeatedly pumped through the cell to remove the residual sample and clean the sample cell (Figure 2C). Note that the rinse syringes are connected to larger solvent reservoirs (e.g., water and ethanol) via pump selector values to ensure that adequate solvent volumes are available to clean the sample cell between measurement runs. Solvent sources, sample sources, and mixed sample containers that contain flammable liquids are positioned in a separate enclosure with no electrical equipment to eliminate all possible ignition sources. In addition, vapor-locking bottle caps are used to minimize flammable vapors and solvent evaporation. Finally, the sample cell is dried with a nitrogen gas stream to remove the residual rinse solvent (Figure 2D). The inlet nitrogen gas pressure to the mixer selector valve is regulated to approximately 2 bar (0.2 MPa, gauge pressure) using a manual pressure regulator located on the nitrogen gas cylinder. Once the sample cell is sufficiently cleaned and dried, a newly mixed sample is injected into the sample cell for the next measurement cycle (repeating the mixing and injection illustrated in the flow diagram in Figure 2A).
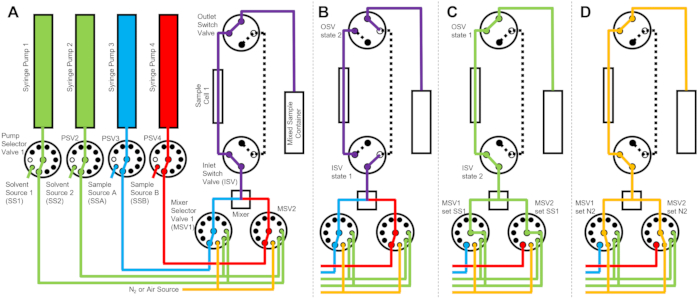
Figure 2: Example flow diagram using one sample cell, two samples mixing, and two rinse solvents for cleaning. (A) Mixing of sample A (blue) and sample B (red), and then flowing the mixed sample (purple) into the sample cell. (B) During data collection, the stopped-flow device state where the ISV and OSV switch valves are closed to isolate the sample cell and prevent back diffusion of the sample during data collection. (C) The cleaning steps where the sample cell is rinsed with rinse solvent from SS1 (green) after data collection. (D) Drying step where the sample cell is dried with nitrogen gas (orange). Abbreviations: PSV = pump selector valve; MSV = mixer selector valve; OSV = outlet switch valve; ISV = inlet switch valve; SS1 = solvent source 1; SSA = sample source A; N2 = nitrogen gas source. Please click here to view a larger version of this figure.
Figure 3 shows flow diagrams for a slightly different version in which the mixing setup is configured with two separate sample cells connected to the same switch valves (Figure 3A). While TR-SANS data are collected in Sample Cell 1, Sample Cell 2 is rinsed (Figure 3B) and dried (Figure 3C). When the data collection is complete for Sample Cell 1, the Inlet Switch Valve directs a newly mixed sample into Sample Cell 2 for data collection (Figure 3D). While TR-SANS data are collected in Sample Cell 2, Sample Cell 1 is rinsed and dried (Figure 3E). This alternating, parallel process between two sample cells minimizes the time between subsequent sample injections and maximizes the use of neutron beam time.
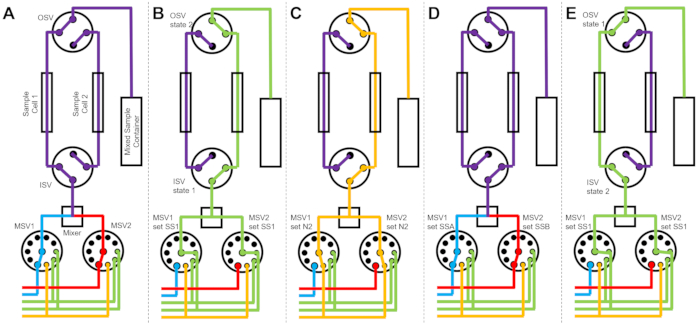
Figure 3: Example flow diagram using a two-sample cells, two samples mixing, and two rinse solvents for cleaning. (A) Mixing sample A (blue) and sample B (red) and then flowing the mixed sample (purple) into sample cell 1. (B) The stopped-flow device state during data collection on sample cell 1 while sample cell 2 is rinsed with solvent from SS1 (green). (C) The stopped-flow device state during data collection on sample cell 1 while sample cell 2 is dried with nitrogen gas (orange). (D) Once data collection of sample cell 1 is complete, a new sample (purple) is immediately mixed and flowed into sample cell 2. (E) The stopped-flow device state during data collection on sample cell 2 while sample cell 1 is rinsed with solvent from SS1 (green). While one sample cell is being measured, the other sample cell is being cleaned and dried. The stopped-flow measurement process alternates between two sample cells to minimize the time between subsequent sample mixing injections. Abbreviations: PSV = pump selector valve; MSV = mixer selector valve; OSV = outlet switch valve; ISV = inlet switch valve; SS1 = solvent source 1; SSA = sample source A; N2 = nitrogen gas source. Please click here to view a larger version of this figure.
A step-by-step protocol is described below for connecting the pumps and tubing lines, priming the system, rinsing and drying the sample cell, and injecting the mixed sample. Although the single-cell configuration is demonstrated for simplicity (Figure 2), the flexible modular setup, protocol, and scripts can be easily modified to implement more syringe pumps, valves, mixers, or sample cell configurations, such as the two-sample cell configuration shown in Figure 3. Representative raw neutron count rate data collected throughout mixing and cleaning injection cycles are shown in Figure 4, while lipid exchange kinetics measured at 3 different temperatures and the extracted normalized scattered intensity corresponding to the fraction of lipids exchanged are shown in Figure 5 and Figure 6, respectively.
The representative neutron data shown here measure lipid exchange kinetics in the presence of methyl-β-cyclodextrin (mβCD), an additive that catalyzes the lipid exchange between vesicles with the exchange rate (ke)66,67. Previous fluorescence studies have shown that ke depends on the mβCD concentration, and the half-life of the exchange process is on the order of minutes68. The present experiments use stopped-flow TR-SANS to measure mβCD-catalyzed lipid exchange between vesicles on the seconds time scale. Two isotopically distinct lipid vesicle populations were prepared; one vesicle population was prepared with tail-hydrogenated dimyristoylphosphatidylcholine (DMPC) lipids (H-lipid vesicles in Figure 5), and the other vesicle population was prepared with tail-deuterated DMPC (DMPC-d54) lipids (D-lipid vesicles in Figure 5). A mole fraction of 0.05 (5 mol%) of charged dimyrisotylphosphatidylglyercol (DMPG) lipid was added to both the DMPC and DMPC-d54 dry lipid powders to promote unilamellar vesicle formation69.
Separate H-vesicle and D-vesicle solutions were prepared by hydrating the respective lipid films in a solvent containing 45% by volume heavy water (D2O) and 55% by volume water (H2O). The D2O/H2O solvent composition was calculated such that the neutron scattering length density (ρ) of the solvent matched a random mixture of the H-lipids and D-lipids (Δρ = ρlipid– ρsolvent = 0). In other words, a randomly mixed H/D-vesicle would be 'invisible' to the neutrons and generate zero coherent neutron scattering. Unilamellar vesicle solutions were prepared by freeze-thawing the solutions five times and then extruding the individual solutions through a polycarbonate filter with 100 nm diameter pores. The vesicle solutions were extruded back and forth between two syringes and the filter for a total of 31 times at 30 °C. Subsequently, a small volume of a concentrated mβCD stock solution prepared in the same D2O/H2O solvent mixture was added to the vesicle solutions. The final lipid concentration was 14 mmol/L (mM) and the mβCD concentration was 30 mM. The individual vesicle solutions were equilibrated for at least 30 min with the added mβCD before the solutions were loaded into the sample syringes in the stopped-flow mixing device.
An example of the measured neutron count rates over multiple injection cycles is shown in Figure 4A. Each cycle consisted of 9 rinsing steps, 1 drying step, and 1 sample injection step. Only count rates measured on the middle detector carriage in the VSANS instrument are presented for clarity. Similar trends were found on the front detector carriage, which corresponded to data collected at larger scattering angles or higher q values. The count rate spiked with each rinsing solvent injection (solvent S1 and solvent S2) and returned to the empty cell baseline counts when the solvent was pushed out of the cell with nitrogen gas and dried. The final cell rinse was with S2, which was ethanol in this example, and the cell was dried a final time with nitrogen gas for 3 min before sample injection. Soon after the sample was injected into the cell, the neutron count rate spiked, and data were collected continuously for 5 min. The representative sample injected in the example data in Figure 4A was a salt buffer background. The fluctuations in measured intensity over time reflect the variations in the background neutron count rate and do not reflect a change in the average sample composition. The complete cycle of rinsing, drying, mixing, and injecting sample was repeated an additional time in the example in Figure 4A, where each cycle lasted for a total of 15 min.
The individual H-labeled and D-labeled lipid vesicle solutions were mixed at time tmix and immediately flowed into the sample cell. The measured neutron count rate spiked and then reached a maximum value when the sample cell was filled at tfill, as shown in Figure 4B. The elapsed time required to fill the sample cell is called the delay time tdelay and is given by tdelay = tfill– tmix. If the void volume (Vvoid) between the mixer and the sample cell is known and the total flow rate (Q) is known, then tdelay can also be calculated as tdelay = Vvoid/Q (see protocol step 4.3.2), which is also the average residence time for the liquid sample to enter the mixer and leave the sample cell. After reaching tfill, the flow was continued at a constant flow rate to ensure that the cell had filled and reached steady-state. The flow was then immediately stopped at tstop. The averaged neutron count rate remained constant as a function of measurement time between tfill and tstop because the flow rate through the sample cell was constant, and therefore, the sample within the neutron beam path was at steady-state. In other words, the data measured at tstop correspond to the sample that has been mixed and evolved by tdelay = tfill– tmix = Vvoid/Q. The binned measurement times ti collected immediately after tstop are the main kinetic data of interest.
In Figure 4C, the neutron counts from the mixed lipid vesicles sample at three different temperatures are plotted as a function of the corrected process time scale (tprocess), which is the process time of interest that has been corrected for the steady-state flow period and delay time. The process time scale was calculated by tprocess = ti– tstop + tdelay, or equivalently, tprocess = ti– tstop + tfill– tmix. SANS data were collected continuously in Figure 4 in so-called event mode. During event mode data collection, each detected neutron event is recorded with a unique time stamp and its x and y location on the two-dimensional neutron detector. Event mode data is then post-processed into the desired time bins (ti) in Figure 4B.
Event mode data within the accessible process time window of interest (i.e., neutron scattering collected at each ti after tstop in Figure 4B) were reconstructed into a two-dimensional (2D) detector image for that time bin using protocols and scripts available in the online open-source repository64. Each 2D detector image was then processed using data reduction routines to subtract the different sources of background, correct for the sample transmission and detector efficiency, and azimuthally integrate the 2D detector data into intensity (I) vs. scattering vector (q) plots65. The data in Figure 5 were binned into equal (3 s) time bins and are representative of the time- and length-scale-dependent information that can be gained from a TR-SANS measurement. Similar to the total raw count rate shown in Figure 4B, the q-dependent intensity I(q) decreases in time as the lipids in the outer leaflet exchange and mix randomly between different vesicles.
Data are presented in Figure 5 for the lipid exchange kinetics measured at 3 different temperatures. Each plot shows the data collected for the first 110 min after mixing. The measured intensity decreases by an order of magnitude at the lowest q-values at 36 °C and 30 °C, which correspond to the lipid fluid phase. Meanwhile, the scattered intensity data change significantly slower with time at 20 °C, indicating that the outer leaflet exchange kinetics are much slower in the lipid gel phase.
The measured scattering intensity, I(q), is related to the SLD contrast as 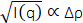 , in which Δρ is the SLD difference between the vesicle and the surrounding solvent. This average SLD contrast is directly related to the relative numbers of H-lipids and D-lipids in a vesicle at any given time. As such, the measured scattering intensity at a given time can be normalized to determine the fraction of the lipid population that has exchanged. This normalization is achieved by collecting two additional measurements, including: (1) the measured intensity I(0) at time t = 0, when there is no lipid exchange between vesicles, and (2) the measured intensity I(∞) at time t = ∞, when all of the lipids have exchanged and the populations have equilibrated. The normalized count rate,
, in which Δρ is the SLD difference between the vesicle and the surrounding solvent. This average SLD contrast is directly related to the relative numbers of H-lipids and D-lipids in a vesicle at any given time. As such, the measured scattering intensity at a given time can be normalized to determine the fraction of the lipid population that has exchanged. This normalization is achieved by collecting two additional measurements, including: (1) the measured intensity I(0) at time t = 0, when there is no lipid exchange between vesicles, and (2) the measured intensity I(∞) at time t = ∞, when all of the lipids have exchanged and the populations have equilibrated. The normalized count rate, 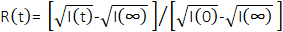 42, is plotted as a function of the process time for the different temperatures in Figure 6. In this example, I(t) is the q-integrated intensity at process time t (background-subtracted intensity integrated as a function of q), I(∞) is the q-integrated intensity at infinite time after all lipids have exchanged (which should be similar to the solvent background scattering), and I(0) is the q-integrated intensity at time t = 0 (with no lipid exchange). I(0) was measured for a mixed sample below the phase transition temperature at 20 °C where the exchange was slow, and I(∞) was measured in a separate sample that had equilibrated for more than 36 h at 40 °C and had and fully exchanged H-lipids and D-lipids.
42, is plotted as a function of the process time for the different temperatures in Figure 6. In this example, I(t) is the q-integrated intensity at process time t (background-subtracted intensity integrated as a function of q), I(∞) is the q-integrated intensity at infinite time after all lipids have exchanged (which should be similar to the solvent background scattering), and I(0) is the q-integrated intensity at time t = 0 (with no lipid exchange). I(0) was measured for a mixed sample below the phase transition temperature at 20 °C where the exchange was slow, and I(∞) was measured in a separate sample that had equilibrated for more than 36 h at 40 °C and had and fully exchanged H-lipids and D-lipids.
The normalized count rate should decay from R(t) = 1 at process time t = 0, to R(t) = 0 at t = ∞, and is directly related to SLD contrast in the sample and therefore the extent of lipid exchange27,28,29,30,31,32,33,34,35. Notice that the earliest measured R(t) data do not begin at R(t)=1 at 30 °C and 36 °C, indicating that a significant amount of lipid exchange occurred during the first 3 s after mixing, which was not experimentally accessible due to the delay time (tdelay = 2.4 s) of the employed stopped-flow mixing protocol. Meanwhile, the measured R(t) at 20 °C remained approximately constant for the first (2-3) minutes. For the lipid exchange kinetics measured at 30 °C and 36 °C, R(t) rapidly decayed to ≈0.5 within 100 s, suggesting the outer leaflet lipids have completely exchanged and equilibrated within minutes. Accordingly, capturing the mβCD-catalyzed lipid exchange was made possible using stopped-flow SANS and would be difficult to capture with manual pipette mixing. Capturing even earlier process time points (t < 3 s) will require a different mixer type with smaller void volume, smaller tubing void volumes, and higher total flow rates to decrease the delay time.
The R(t) continued to decay at longer times as the lipids flip-flop between the inner and outer leaflets. TR-SANS data for the slower flip-flop process (t > 5 min) can be collected with discrete samples mixed by hand and loaded into standard SANS sample cells, as the manual pipette mixing method takes approximately 5 min. Similarly, the lipid exchange at 20 °C in the lipid gel phase was slow enough to be mixed and measured discretely, and this measurement did not necessarily need to be studied with the stopped-flow mixing method. Measurements of kinetics processes on time scales of several minutes to hours are more efficient when the samples are mixed by manual pipette mixing. However, kinetic processes on time scales less than minutes will require this stopped-flow mixing and TR-SANS measurement procedure.
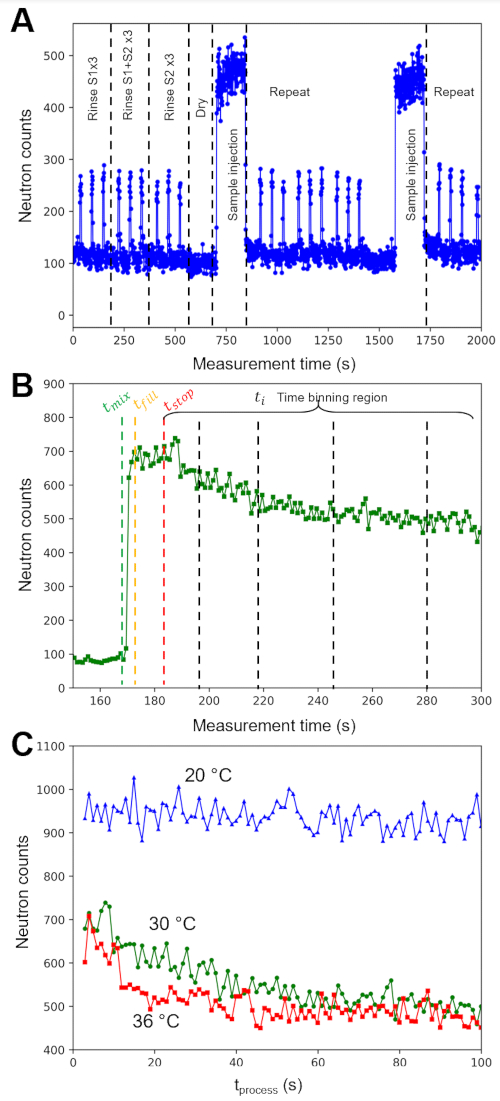
Figure 4: Representative raw neutron count rate data collected throughout mixing and cleaning injection cycles. (A) Example of the neutron count rate (middle detector) as a function of time during repeated rinsing sequences, drying sequence, and mixed sample injection sequence over multiple cycles. The sample in (A) was a salt buffer background, and the changes in intensity over time reflect the variations in the background count rate, not a change in the sample. (B) Monitor-normalized neutron count rate as a function of the experiment time after the injection of mixed H-lipid and D-lipid vesicles at 30 °C. The vertical dashed lines indicate the mixing start time (tmix), the fill time (tfill), the flow stop time (tstop), and time binning region (ti). The decay in count rate after tstop is due to loss of contrast in the sample as the lipid exchange between vesicles. (C) Monitor normalized neutron count rates as a function of exchange process time for the first 100 s of the experiment at (blue) 20 °C, (green) 30 °C, and (red) 36 °C. The event mode data are processed into 1 s bins. Abbreviations: S1 = solvent 1; S2 = solvent 2; tmix = experiment time at which the sample solutions are mixed; tfill = experiment time at which the sample cell is filled; tstop = experiment time at which the flow is stopped; tprocess = calculated kinetic process time of interest. Please click here to view a larger version of this figure.
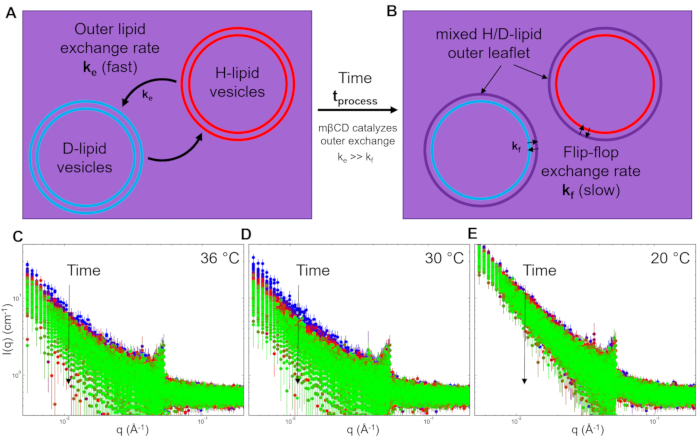
Figure 5: Illustration of catalyzed exchange of the outer layer of lipid vesicles and corresponding changes in the scattered intensity (I) as a function of the scattering vector (q) at various temperatures. Schematic showing lipid exchange between H-lipid and D-lipid vesicles after (A) initial mixing at t = 0 and (B) exchange of the outer layer catalyzed by methyl-β-cyclodextrin (mβCD). Reduced neutron scattering intensity as a function of scattering wave vector q. Stopped-flow mixing experiments and VSANS measurements were repeated at (C) 36 °C, (D) 30 °C, and (E) 20 °C. The presented data were binned into 3 s intervals over the first 10 min after mixing at each specified temperature. Error bars are the propagated uncertainty from the counting statistics and represent one standard deviation. Abbreviations: ke = rate constant for inter-vesicle lipid exchange; mβCD = methyl-β-cyclodextrin; kf = rate constant for inter-leaflet lipid exchange, also referred to as lipid flip-flop; l(q) = measured SANS intensity with units of cm-1 ; q = scattering vector. Please click here to view a larger version of this figure.
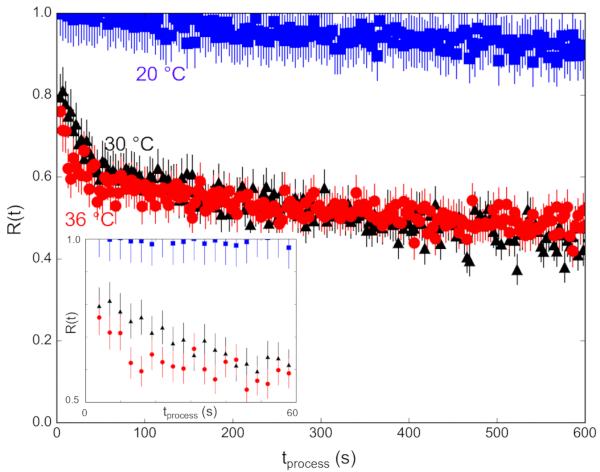
Figure 6: Normalized scattered intensity corresponding to the fraction of lipids exchanged that can be modeled to extract rate constants for the kinetic processes of interest. (A) Lipid exchange between the outer leaflet of the vesicles occurring at time scales between 3 s and 600 s measured at (blue) 20 °C, (black) 30 °C, and (red) 36 °C. The inset in the figure zooms in on the first 60 s of the kinetic process. Error bars are the propagated uncertainty from the numerical integration of the scattering intensities and represent one standard deviation. Abbreviations: R(t) = normalized scattered intensity; tprocess = corrected process time of interest. Please click here to view a larger version of this figure.
