Оценка in vitro и in vivo фотоконтролируемых биологически активных соединений - потенциальных кандидатов на лекарства для фотофармакологии рака
Summary
Этот протокол представляет собой набор экспериментов, принятых для оценки фотопереключаемых противоопухолевых пептидов, которые могут быть использованы в доклиническом скрининге таких соединений. Это включает в себя оценку цитотоксичности в 2D и 3D клеточных культурах, оценку эффективности фотоизомеризации ex vivo (модельной ткани) и эффективности in vivo .
Abstract
Фотоконтролируемые, биологически активные соединения являются новым классом «умных» кандидатов на лекарства. Они обеспечивают дополнительную безопасность при системной химиотерапии благодаря их точной пространственно-временной активации, направляя доброкачественный, неионизируемый свет в определенное место в организме пациента. В данной работе представлен комплекс методов оценки in vitro потенции и эффективности ex vivo фотоактивации фотоконтролируемых биологически активных соединений, а также эффективности in vivo на ранних стадиях разработки лекарственных средств. Методика применяется к противоопухолевым цитотоксическим пептидам, а именно к диарилетеносодержащим аналогам известного антибиотика грамицидина С. Эксперименты проводятся с использованием 2D (адгезивные клетки) и 3D (сфероиды) клеточных культур линии раковых клеток (карцинома легкого Льюиса, LLC), суррогатов живых тканей (фарш из свинины) и модели рака аллотрансплантата (подкожная LLC) у иммунокомпетентных мышей. Выбор наиболее эффективных соединений и оценка реалистичных фототерапевтических окон выполняются с помощью автоматизированной флуоресцентной микроскопии. Эффективность фотоактивации при различных режимах освещения определяется на разной глубине в модельной ткани, а оптимальная дозировка света применяется в заключительном терапевтическом эксперименте in vivo.
Introduction
Фотоконтролируемые биологически активные соединения появились в последние десятилетия в качестве перспективного компонента безопасной химиотерапии заболеваний человека и специфического искоренения злокачественных солидных опухолей1. Эти соединения содержат обратимо фотоизомеризуемые фрагменты (молекулярные фотопереключатели) и могут переключаться между неактивными и активными фотоизомерами при облучении светом разных длин волн.
По сравнению со своими нефотоконтролируемыми аналогами, фотоконтролируемые препараты могут быть более безопасными, поскольку они могут систематически вводиться в организм пациента в менее активных и по существу нетоксичных формах, а затем активируются светом только там, где это необходимо, например, в опухолях, язвах и ранах. Несмотря на то, что в недавних научных работах 2,3,4,5,6,7 можно найти множество захватывающих демонстраций таких молекулярных прототипов лекарств, области клинической фотофармакологии – применения одобренных комбинаций лекарств / медицинских устройств / заболеваний – не существует. Фотофармакология все еще находится на стадии открытия лекарств, и систематические доклинические исследования неизвестны.
Совсем недавно мы продемонстрировали преимущество безопасности in vivo для некоторых фотоконтролируемых противоопухолевых пептидов, а именно аналогов пептидного антибиотика грамицидина S8. Эти фотоуправляемые производные содержат фотопереключатель с диарилетеном (DAE), который претерпевает обратимые фотоиндуцированные преобразования между так называемыми фотоформами, генерируемыми красным светом, и фотоформами, генерируемыми УФ-излучением, «замкнутыми кольцами» (проиллюстрировано на рисунке 1 для одного из производных, соединения LMB002).
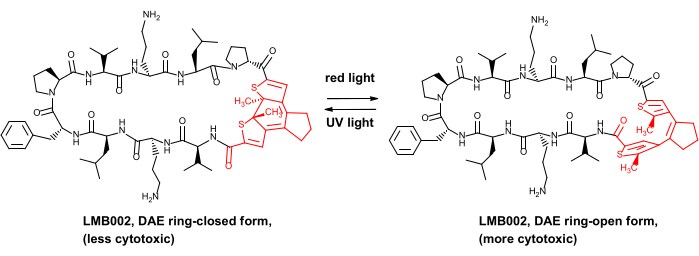
Рисунок 1: Фотоуправляемый цитотоксический пептид LMB002 и его фотоизомеризация. Фрагмент дневника показан красным цветом. Аббревиатура: DAE = diarylethene. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
Для нахождения попаданий и оптимизации попадания в лид часто требуется in vitro и in vivo скрининг соответствующих библиотек соединений 9,10. Здесь мы демонстрируем методологию, подходящую для систематического высокопроизводительного скрининга цитотоксичности фотоконтролируемых соединений. Мы также определяем эффективность фотоизомеризации, оцениваем дозу света в модельных тканях и оцениваем эффективность in vivo наиболее эффективных кандидатов. Этот подход соответствует соображениям биоэтики и ухода за животными.
В этой работе традиционные доклинические методы модифицируются, чтобы избежать неконтролируемой фотоизомеризации испытуемых соединений. Общая цель применения этих модифицированных методов состоит в том, чтобы разработать общую стратегию, которая является простой и быстрой и дает статистически значимые данные для надежного сравнения активности in vitro и рационализации тестирования эффективности фотопереключаемых соединений in vivo для идентификации свинца и дальнейшей разработки.
Стратегия состоит из трех последовательных шагов. Первый этап включает определение IC 50 (кажущаяся50% жизнеспособность клеток) в серийных разведениях для активных и неактивных фотоформ выбранных фотоконтролируемых биологически активных соединений с использованием двумерных (2D, монослойных) и трехмерных (3D, сфероидных) клеточных культур и конфокальной высокопроизводительной автоматизированной флуоресцентной микроскопии. Фототерапевтические окна сравниваются с разницей IC50 между двумя фотоформами, и отбираются наиболее эффективные кандидаты. Нет особых преимуществ в оценке токсичности с помощью автоматизированной микроскопии и других платформ (анализов) скрининга цитотоксичности11; Более сложные клеточные моделиопухолей 12 могут быть легко реализованы на этом этапе.
Для соединений, выбранных на этапе 1, второй этап заключается в реалистичной оценке их эффективности фотопереключения внутри тканей в зависимости от глубины от облученной поверхности ткани путем количественной оценки эффективности фотопереключения менее активных фотоформ в тканевом суррогате с использованием высокоэффективной жидкостной хроматографии (ВЭЖХ) облученных образцов с УФ-детектированием. In vivo эффективность фотопереключения можно было бы изучить, но мы предлагаем использовать простой суррогат ткани – фарш из свинины. Мы проверили обоснованность такого подхода. Мы измерили конверсию наших фотопереключаемых соединений in vivo на модели рака мышей и наблюдали примерно такую же фотоконверсию на глубине, измеренной в предыдущих экспериментах с мышами8. Может быть использована любая подходящая альтернативная искусственная ткань13, 3D-биопечатная ткань/орган14, биопсийные материалы или другой освобожденный материал животного происхождения. Тем не менее, эта установка является хорошим компромиссом, поскольку она экономична, быстра и этична.
Третьим шагом является определение противоопухолевой эффективности in vivo в модели рака мышей. Для эксперимента отобраны соединения, демонстрирующие превосходные характеристики в экспериментах in vitro и эффективно фотопереключающиеся на глубине не менее 1-1,5 см в модельных тканях.
Этот протокол может быть применен к соединениям, обладающим различными типами фотопереключателей, при условии, что их фотоформы (или их фотостационарные состояния, PSS) стабильны в течение разумного времени (несколько дней или дольше). В качестве иллюстрации используется ранее описанный производный от DAE LMB00215. Фотоформы LMB002 термически стабильны и могут храниться при температуре -20 °C не менее года без существенной деградации. Для демонстрации in vitro и in vivo выбираются клетки карциномы легкого Льюиса (LLC), но никаких ограничений на тип клеток не накладывается. Клетки LLC являются адгезивными, легко культивируемыми в 3D и используются для получения опухолей (как описано в ссылке16). В естественных условиях Клетки LLC используются для моделирования метастатических процессов и могут легко генерировать солидные опухоли у иммунокомпетентных мышей после подкожной инъекции. Эта методология in vivo может быть универсально применена к другим моделям рака17,18. Ниже приводится подробное описание осуществления этой стратегии.
Protocol
Representative Results
Discussion
Фотоконтролируемые соединения являются беспрецедентными в разработке лекарств; Однако не было установлено никаких методов для их доклинической и клинической оценки. Ближайший аналог монотерапии, фотодинамическая терапия (ФДТ), является методом лечения для клинического применения, принятым во многих странах против рака, и находится в разработке для других показаний19,20. Подобно фотофармакологии, ФДТ также основана на использовании света для активации биологически активного вещества (синглетного кислорода). Таким образом, некоторые экспериментальные методы, используемые для доклинических и клинических исследований при ФДТ, могут быть приняты для фотофармакологии. Например, источники света, подходы к подаче света и медицинские устройства хорошо разработаны и одобрены для ФДТ; Они могут быть непосредственно использованы для оценки фотоконтролируемых препаратов. Однако ФДТ и фотофармакология имеют много отличий друг от друга4, что обосновывает необходимость установления специфических методов для последней.
Во-первых, неактивированное вещество в ФДТ (кислороде) всегда присутствует в живых тканях в нетоксичных концентрациях. Напротив, неактивированные фотоконтролируемые биологически активные соединения могут иметь остаточную активность и нежелательную токсичность. Следовательно, идеальные фотофармакологические препараты должны иметь минимизированную биологическую активность в вводимой форме и должны быть высокоактивными в своей светогенерируемой форме, «фототерапевтическое окно»21 должно быть как можно больше. Поиск попадания и выполнение оптимизации «попадание в свинец» требует идентификации подходящих соединений и скрининга относительно больших библиотек уже на ранних стадиях разработки лекарств. Здесь мы предложили автоматизированную высокопроизводительную конфокальную флуоресцентную микроскопию для идентификации эффективных фотокоммутационных соединений.
Выбранный метод оценки цитотоксичности позволяет легко реализовать наиболее критическое требование – поддержание PSS или стабильности видимого светочувствительного фотоизомера. Это связано с тем, что при его реализации воздействие света сводится к минимуму. Следовательно, при выборе альтернативных методов следует отдавать предпочтение автоматизированным. Такой подход надежен и информативен. Использование 3D-клеточных культур (сфероидов) на этом этапе обеспечивает целостное понимание реакции клетки на лечение в более реалистичном тканеподобном микроокружении. Кроме того, ценная информация о механизме действия соединений может быть получена с помощью микроскопии в качестве прямого метода. Конфокальная флуоресцентная микроскопия с надлежащим протоколом окрашивания позволяет визуально оценить морфологию клеток и сфероидов; Также могут быть обнаружены важные детали гибели клеток и изменений внутри клеток.
Во-вторых, легкое нанесение требует тщательного подбора легкой дозировки. При ФДТ передозировка света чрезвычайно вредна для тканей22. Фотофармакологическая терапия может быть полезна при чрезмерном облучении светом. Верхний предел действия активированного вещества определяется введенной дозой неактивированного вещества и его фармакокинетикой. Тем не менее, легкая дозировка по-прежнему является проблемой в фотофармакологии. Следует позаботиться о том, чтобы плотность мощности облучения и время воздействия были не меньше , чем требуется для терапии. В принципе, генерацию активированного вещества можно контролировать in vivo. Однако из соображений биоэтики мы предложили эксперимент с модельной тканью (свежим фаршем), смешанной с неактивированным соединением15. Этот эксперимент прост и может быть модифицирован для использования различных источников света. Он также может быть адаптирован для фотофизической оценки дозировки света и измерения тепловых воздействий. Опять же, используя модельные ткани, можно свести к минимуму воздействие света, по сравнению, например, с более точным определением эффективности фотопереключения в условиях in vivo , альтернативой, которую всегда может быть интересно рассмотреть.
Наконец, соединения, которые демонстрируют превосходные характеристики в скринингах токсичности in vitro и эффективно переключаются на глубину не менее 1-1,5 см в модельной ткани, могут быть выбраны для дорогостоящих, трудоемких и длительных исследований in vivo . В этом протоколе мы использовали ту же клеточную линию (LLC), что и в оценке in vitro , для создания модели рака аллотрансплантата. Динамика роста опухоли, смертность и количество метастазов являются параметрами, наиболее подходящими для оценки противоопухолевой эффективности. По сравнению с обычной химиотерапией, в фотофармакологическом лечении применяется дополнительный фактор – свет. Следовательно, необходимы две группы контрольных животных: одна, которая получает только транспортное средство, и другая, которая получает транспортное средство и облучение. Эта установка позволяет оценить влияние света на измеряемые параметры. В нашем эксперименте животные двух экспериментальных групп получали неактивированное соединение, а опухоли мышей в одной группе облучали. Режим облучения был идентичен для контрольной и лечебной групп. Сравнение с эталонной химиотерапией на данном этапе не требуется, поскольку основная цель эксперимента – продемонстрировать комбинированный эффект применения света и соединения. Наиболее эффективные соединения, демонстрирующие этот эффект, затем могут быть выбраны для дальнейшего изучения их токсичности in vivo и сравнения с контрольными показателями для принятия важных решений по их разработке. Технически эксперимент in vivo , который мы описываем, может быть легко адаптирован к фармакокинетическим или фармакодинамическим исследованиям, например, соединения, которое уже выбрано в качестве лекарственного средства.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Авторы признают финансирование ЕС программой H2020-MSCA-RISE через проекты PELICO (#690973) и ALISE (#101007256). Эта работа была поддержана DFG-GRK 2039 (SA, TS и ASU), программой NACIP Общества им. Гельмгольца (SA и ASU) и VIP+ BMBF (OB и ASU). Мы благодарим доктора Сергея Конева из Технологического института Карлсруэ, который синтезировал соединение LMB002, очистил его и любезно предоставил соединение для исследования. Авторы также благодарят Чуприну Максима, который снял и собрал видео в Украине, и всех отважных защитников Украины, которые сделали возможной экспериментальную работу, написание и съемку этой публикации.
Materials
| Agilent 1100 Series capillary LC system | ALSI-Chrom (Agilent distributor) | – | |
| ATCC CRL-1642, LL/2 (LLC1) Lewis lung carcinoma cell line | ECACC | 90020104 | |
| C57BL/6NCrl mice, female, inbred | Charles River | Strain code: 027 | |
| CelCulture, CO2 incubator | Esco Micro | CCL-170B | |
| Corning Matrigel Basement membrane matrix | Merck | CLS354234 | |
| Corning, 384- well spheroid microplates | Merck | CLS3830 | |
| Fetal bovine serum | Merck | F7524 | |
| Gibco, DPBS | Thermo Fisher Scientific | 21600044 | |
| Gramicidin S | Lumobiotics | Custom synthesis | |
| HyClone, DMEM/high glucose | Cytiva | SH30003.04 | |
| IN Cell Analyzer 6500HS, imaging system | Cytiva | 29240358 | |
| Invitrogen, Calcein AM | Thermo Fisher Scientific | C1430 | |
| Isoflurane anesthesia machine | ASA | S/N ASA 1305 | |
| L-glutamine, 200 mM solution | Merck | G7513 | |
| LIKA-surgeon, diode surgery laser | Fotonika plus | – | |
| LMB002 | Lumobiotics | Custom synthesis | |
| Penicillin–Streptomycin, solution stabilized | Merck | P4333 | |
| PhenoPlate, 96-well plates | PerkinElmer | 6055302 | |
| Photometer PCE-LED 20 | PCE Instruments | PCE-LED 20 | |
| Thermo Scientific, Hoechst 33342 | Thermo Fisher Scientific | 62249 | |
| Thermo Scientific, Propidium iodide | Thermo Fisher Scientific | J66764-MC | |
| Trypan blue, 0.4% solution | Merck | T8154 | |
| Trypsin–EDTA, 10 x solution | Merck | T4174 | |
| UltraCruz Cell culture flasks with vented caps, 75 cm2 | Santa Cruz Biotechnology | sc-200263 | |
| UltraCruz, bottle top filters, PES, 0.22 μm | Santa Cruz Biotechnology | sc-360882 | |
| Vydac 218TP, C18 HPLC column (4.6 mm × 250 mm, 5 µm) | Altmann Analytik (Avantor distributor) | GR5103827 |
References
- Fuchter, M. J. On the promise of photopharmacology using photoswitches: a medicinal chemist’s perspective. Journal of Medicinal Chemistry. 63 (20), 11436-11447 (2020).
- Volarić, J., Szymanski, W., Simeth, N. A., Feringa, B. L. Molecular photoswitches in aqueous environments. Chemical Society Reviews. 50, 12377-12449 (2021).
- Paoletti, P., Ellis-Davies, G. C. R., Mourot, A. Optical control of neuronal ion channels and receptors. Nature Reviews Neuroscience. 20, 514-532 (2019).
- Hüll, K., Morstein, J., Trauner, D. In Vivo Photopharmacology. Chemical Reviews. 118 (21), 10710-10747 (2018).
- Ma, X., et al. In vivo photopharmacology with a caged mu opioid receptor agonist drives rapid changes in behavior. Nature Methods. 20, 682-685 (2023).
- Sarabando, S. N., Palmeira, A., Sousa, M. E., Faustino, M. A. F., Monteiro, C. J. P. Photomodulation Approaches to Overcome Antimicrobial Resistance. Pharmaceuticals. 16 (5), 682 (2023).
- Kolarski, D., Szymanski, W., Feringa, B. L., Hirota, T., Hatori, M., Panda, S. Chronophotopharmacology: Methodology for high spatiotemporal control over the circadian rhythm with light. Neuromethods. 186, (2022).
- Babii, O., et al. Peptide drugs for photopharmacology: how much of a safety advantage can be gained by photocontrol. Future Drug Discovery. 2 (1), FDD28 (2020).
- Davis, A. M., Keeling, D. J., Steele, J., Tomkinson, N. P., Tinker, A. C. Components of successful lead generation. Current Topics in Medicinal Chemistry. 5 (4), 421-439 (2005).
- Balani, S. K., Miwa, G. T., Gan, L., Wu, J., Lee, F. W. Strategy of utilizing in vitro and in vivo adme tools for lead optimization and drug candidate selection. Current Topics in Medicinal Chemistry. 5 (11), 1033-1038 (2005).
- Kleijn, A., et al. A Systematic comparison identifies an ATP-based viability assay as most suitable read-out for drug screening in glioma stem-like cells. Stem Cells International. 2016, (2016).
- Rodrigues, J., Heinrich, M. A., Teixeira, L. M., Prakash, J. 3D in vitro model revolution: unveiling tumor-stroma interactions. Trends in Cancer. 7 (3), 249-264 (2021).
- Sittinger, M., et al. Tissue engineering and autologous transplant formation: practical approaches with resorbable biomaterials and new cell culture techniques. Biomaterials. 17 (3), 237-242 (1996).
- Matai, I., Kaur, G., Seyedsalehi, A., McClinton, A., Laurencin, C. T. Progress in 3D bioprinting technology for tissue/organ regenerative engineering. Biomaterials. 226, 119536 (2020).
- Babii, O., et al. Direct photocontrol of peptidomimetics: an alternative to oxygen-dependent photodynamic cancer therapy. Angewandte Chemie International Edition. 55 (18), 5493-5496 (2016).
- De Ridder, K., et al. Novel 3D lung tumor spheroids for oncoimmunological assays. Advanced NanoBiomed Research. 2 (4), 2100124 (2022).
- Pauli, C., et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discovery. 7 (5), 462-477 (2017).
- Van Straten, D., Mashayekhi, V., De Bruijn, H. S., Oliveira, S., Robinson, D. J. Oncologic photodynamic therapy: basic principles, current clinical status and future directions. Cancers. 9 (2), 19 (2017).
- Li, X., Kwon, N., Guo, T., Liu, Z., Yoon, J. Innovative strategies for hypoxic-tumor photodynamic therapy. Angewandte Chemie International Edition. 57 (36), 11522-11531 (2018).
- Hull, K., Morstein, J., Trauner, D. In vivo photopharmacology. Chemical Reviews. 118 (21), 10710-10747 (2018).
- Babii, O., et al. Structure-activity relationships of photoswitchable diarylethene-based β-hairpin peptides as membranolytic antimicrobial and anticancer agents. Journal of Medicinal Chemistry. 61 (23), 10793-10813 (2018).
- Heckl, C., Aumiller, M., Rühm, A., Sroka, R., Stepp, H. Fluorescence and treatment light monitoring for interstitial photodynamic therapy. Photochemistry and Photobiology. 96 (2), 388-396 (2020).
