Expansion of NPCs (Figure 1)
When neurospheres are broken down to a single cell suspension by trituration, the pipette tip needs to touch the bottom of the tube so that there is some resistance when the suspension is pipetted up and down. The number of times of trituration will vary between individuals and needs to be decided by trial and error by examining the resulting cell suspension under a microscope. It is critical to provide sufficient density of cell suspension because the proliferation will be at a faster rate when the NPCs come in close contact. If NPCs do not grow, the cell density should be increased by reducing the number of flasks. We have been able to expand the neurospheres for up to 10-15 passages. Thus abundant supply of human NPCs can be generated, reducing the use of human fetal tissue. For continued supply of NPCs and neurons, it is desirable to expand them initially for 3-4 passages and subsequently, half of the NPCs can be expanded as neurospheres and the other half can be differentiated for ongoing experiments. Alternatively, NPCs can be also expanded for more passages, frozen in liquid nitrogen as in the case of cell lines to generate larger supply of stocks, and revived when needed. It is also desirable to perform experiments with neurons derived from different batches of NPCs.
Neuronal Differentiation of NPCs (Figure 2)
NPCs are multipotent cells and can be differentiated into neurons, astrocytes or oligodendrocytes depending on the culture conditions. Our differentiation protocol involved seeding of 4-day old neurospheres in coated dishes (Figure 2A). We observed that it is essential to seed sufficient density of neurospheres so that the differentiating neurons spread around and form a dense network of neuronal processes. Images after 1 (Figure 2B) and 4 days (Figure 2C) of seeding are shown. Neuron-rich cultures yielding 80-90% neurons and 10-15% astrocytes, were obtained after differentiation of NPCs in the presence of a combination of NGF, BDNF, DBC and retinoic acid for two weeks (Figure 2D). To the best of our knowledge, the protocol described in this study for neuronal differentiation has not been reported previously by others. In areas of low density, NPCs differentiated into astrocytes (Figure 2E). To obtain an astrocyte-rich culture, NPCs can be cultured in the presence of CNTF (10 ng/ml) and in the absence of NGF, BDNF, DBC and retinoic acid. Thus the neuron-astrocyte composition can be manipulated during differentiation depending on the objective of the experiment.
Characterization of Differentiated Neurons (Figure 3)
To further characterize these differentiated NPCs, we performed dual immunostaining for neuronal markers with polyclonal and monoclonal antibodies, followed by exposure to rabbit and mouse secondary antibodies linked to Cy3 (red) and FITC (green) respectively. The following neuronal markers were detected: Figure 3A: NeuN and synapsin. Figure 3B: Acetyl cholinesterase (Ache) and synaptophysin. Figure 3C: BDNF and GAP43. Thus, the differentiated NPCs have the characteristics of primary neurons. In addition, astrocyte markers STAT3 and GFAP were detected in a small population of cells (Figure 3A). Therefore additional methods are needed for neuron-specific gene expression analysis.
Laser Capture Microdissection (LCM) of Neurons (Figure 4)
Although LCM can be used for isolation of cells from a culture dish, our initial attempts of LCM with neurons failed because they are firmly attached to the coated dishes. To overcome this problem, we differentiated NPCs on slides with PEN membrane inserts coated with poly-L-lysine and mouse laminin. The slide was placed inside a 100 mm cell culture dish (Figure 4A). The arrangements of the PEN membrane slide and LCM caps are shown in Figure 4B. Cultures were stained with HistoGen stain (Arcturus) to visualize the cells (Figure 4C). The slide was placed in the microscope in the Arcturus Veritas instrument. The neurons to be captured were marked with drawing tools (Figure 4D). CapSure HS Caps were placed on the PEN membrane. Using a combination of UV laser cutting and IR laser capture, the marked areas were collected onto the cap. The captured neurons are shown on the cap at low (Figure 4F) and high (Figure 4F) magnification.
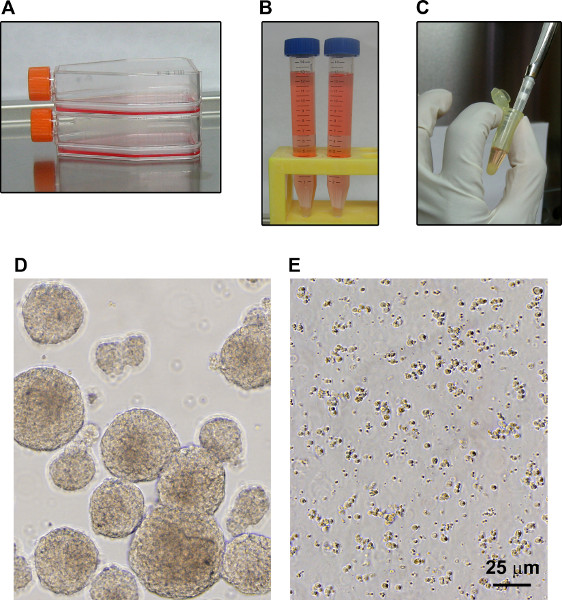
Figure 1. Expansion of human NPCs. A. NPCs were cultured in suspension in T75 flasks as neurospheres. B. When the neurospheres reached the size of 300-500 μm, they were transferred to 15 ml tubes and centrifuged at 1,000 RPM for 5 min. C. The supernatant was discarded and the cell pellet in 200 μl medium was transferred to Eppendorf tubes and triturated. D. Neurospheres ready to be split are shown. E. Broken neurospheres following trituration are shown.
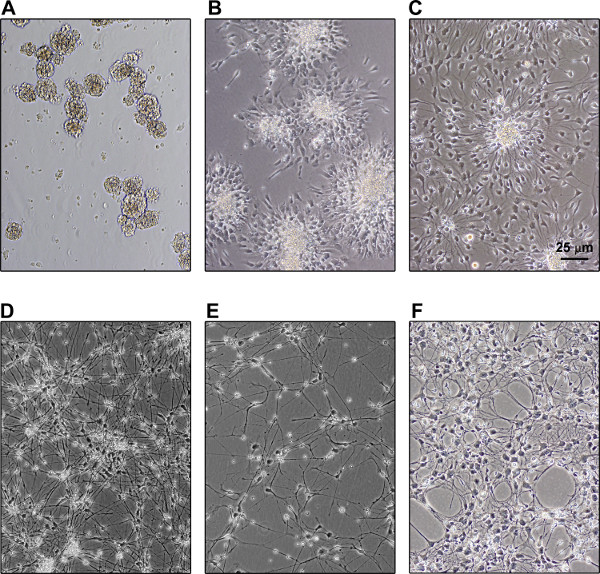
Figure 2. Differentiation of human NPCs. A. Neurospheres were broken by trituration and cultured for four days to generate new neurospheres. B. Four day-old neurospheres were seeded in dishes coated with poly-L-lysine and mouse laminin. C. Differentiation of NPCs was observed in the presence of NGF, BDNF, DBC and retinoic acid in three days after seeding. D. Neuron-rich culture was obtained after two weeks. E. NPCs differentiated into astrocytes in areas of low density. F. NPCs differentiated into an astrocyte-rich culture in the presence of CNTF (10 ng/ml) and in the absence of NGF, BDNF, DBC and retinoic acid.
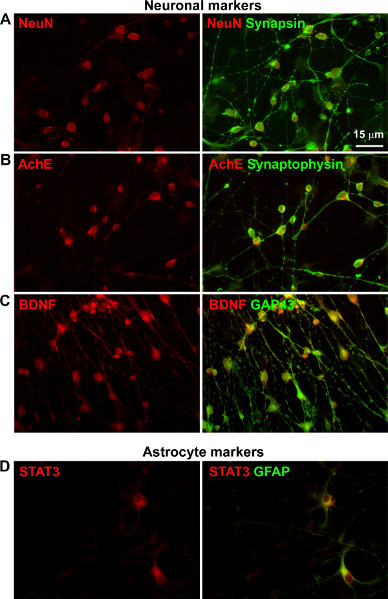
Figure 3. Neuronal and astrocyte markers in differentiated human NPCs. NPCs differentiated for two weeks were fixed and dual immunostained for indicated targets with polyclonal and monoclonal antibodies, followed by exposure to rabbit and mouse secondary antibodies linked to Cy3 (red) and FITC (green) respectively. The following neuronal markers were detected: A. NeuN and synapsin. B. Acetyl cholinesterase (AchE) and synaptophysin. C. BDNF and GAP43. In addition, the following astrocyte markers were detected: D. STAT3 and GFAP.
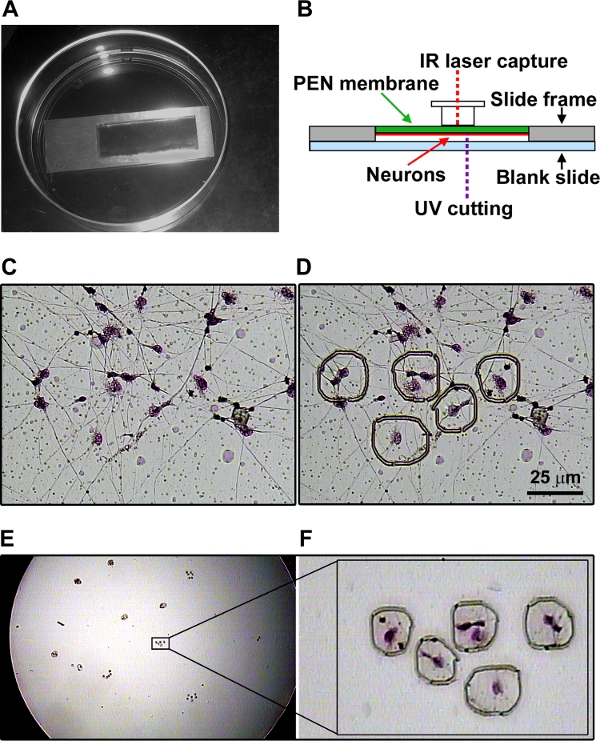
Figure 4. Laser capture microdissection of neurons. A. NPCs were differentiated on a PEN membrane slide coated with poly L lysine and mouse laminin. The slide was placed inside a 100 mm cell culture dish. B. The arrangements of PEN membrane slide and LCM cap in the Arcturus Veritas instrument are shown. C. Cultures were stained with HistoGen stain (Arcturus) to visualize the cells. D. The neurons to be captured were marked with drawing tools. The captured neurons are shown on the cap at low (E) and high (F) magnification. Click here to view larger figure.
