Representative results for green gel electrophoresis are presented in Figure 1. Lane 1 provides an example of ideal results for green gel electrophoresis of spinach thylakoids, in which a maximum number of clear, sharp green bands are visible. These results are somewhat atypical, in part because not all of the bands seen in lane 1 are normally present in a given sample. Additional sample cleanup, in the form of chloroplast isolation before thylakoid solubilization, and gradient gel electrophoresis (4-7% acrylamide) are also normally necessary to achieve optimal results. Lanes 2 and 3 present more typical results achieved using the protocol detailed here in conjunction with a 5% non-gradient gel. Lanes 4, 5, and 6 provide an example of poor results due to increasing degrees of under-solubilization of the thylakoid sample. Lane 7 provides an example of typical results achieved with Arabidopsis thylakoids instead of spinach. Note that for Arabidopsis the megacomplex bands at the top of the gel tend to be poorly resolved compared to those in spinach.
A graphic depiction of the TCSPC setup used for collecting data from native green gel bands is shown in Figure 2. Figure 3 shows a typical workflow for beginning analysis after TCSPC data has been collected as described in step 10. When TCSPC data is collected, each curve at a given wavelength represents an arbitrary number of data points, or "counts". When these curves are overlaid with one another, as shown in Figure 3A, the curves cannot be compared with one another directly because they are not represented at the same scale and may not all be registered to the same starting time point. The first step in data analysis is therefore to compare each curve to its corresponding instrument response function (IRF). The peak of the IRF for a given curve is set to time t = 0, and the leading edge of the corresponding fluorescence decay curve is set to overlap the leading edge of the IRF, as shown in Figure 3B. This will set all curves to the same time register for later analysis.
While it is not necessary for the construction of DAS, normalizing all curves to the same peak height at this point allows a useful comparison between curves to be made as a first analysis of the data. In Figure 3C, the same decay curves for LHCII presented in Figure 3A are shown after time registration, as in Figure 3B, and peak height normalization. As seen in Figure 3D, peak-normalized decay curves from different complexes can then be overlaid with one another at a given wavelength, allowing the differences in behavior between the complexes to be visualized. For example, LHCII has a characteristically long-lived fluorescence that decays slowly, whereas PSI fluorescence is strongly quenched, decaying very rapidly. The Band 5 fluorescence decay curve provides an interesting example of very suggestive data, in part because it is clearly biphasic. The initial fluorescence decay curve for the Band 5 complex follows the same rate as PSI for approximately 500 ps yet decays even more rapidly thereafter. Intriguing results of this kind can be analyzed in further detail by constructing decay-associated spectra (DAS).
In Figure 4, representative DAS waterfall plots are shown for LHCII and the Band 5 complex. Construction of DAS first requires that the decay curves for a given complex be tail-matched to the room temperature fluorescence spectrum for the complex, as described in step 11.2. The results of tail-matching decay curves for LHCII are shown in Figure 4A. DAS are then constructed from these curves as described in step 11.3. DAS for LHCII were constructed from the decay curves shown in Figure 4A and the results are presented in Figure 4B. DAS between 0 and 100 ps were omitted for clarity, and only presented every 100 ps thereafter due to the characteristically slow decay for isolated LHCII. The DAS for LHCII is notable for the lack of dynamic features, and the shape of the LHCII fluorescence spectrum remains the same as the signal decays over time. The decay of the fluorescence spectrum is also delayed, requiring 100 ps to reach maximum fluorescence. This suggests, as would be expected for isolated light harvesting protein complexes, that energy is not transferred between energetically distinct pigments within the complex as the fluorescence decays. The exception to this is the shift in the spectrum occurring during the first 100 ps, presumably due to the initial redistribution of excitation energy throughout the complex.
DAS for the Band 5 complex are shown in Figure 4C, constructed in a similar fashion to that shown for LHCII. Band 5 provides an instructive contrast to LHCII in several ways. Compared to LHCII, fluorescence from Band 5 decays much more rapidly, reaching maximum intensity in only 30 ps and decaying to less than 20% of initial intensity after 500 ps. The fluorescence spectrum of the Band 5 complex also exhibits a number of interesting dynamics as the emission decays. Comparing the spectra at 0 and 60 ps clearly shows an increase in fluorescence at 720 nm at the expense of fluorescence at 680 and 710 nm. Thereafter, the peak at 680 nm shifts towards 690 nm and broadens, while the peak at 720 nm shifts back toward 710 nm (e.g., compare 60 ps to 500 ps). Data of this type helps rule out the presence of unconnected LHCII antenna proteins while providing evidence for energy transfer from LHCII to the PSI antenna and eventually the PSI core.
These dynamics also suggest that there are likely to be unresolved peaks around 680 nm, 690 nm, 710 nm, and 720 nm. This data therefore provides an example where spectral features could be better resolved by collecting TCSPC data at closer intervals (e.g. every 5 nm rather than every 10 nm). Even in the absence of higher spectral resolution, however, the DAS for the Band 5 complex are an example of evidence for energy transfer between multiple fluorescing species within a complex. There also appears to be a rapidly decaying peak above 740 nm, suggesting that data should also be collected over a broader spectrum to include further spectral features.

Figure 1: Representative green gel results. Green gel bands subjected to TCSPC analysis are labeled PSI-LHCII (photosystem I LHCII megacomplexes), PSI (photosystem I LHCI), Band 5 (PSI-LHCII complex), PSII-LHCII (photosystem II and associated light-harvesting complex II), PSII (photosystem II core complex), and LHCII (light-harvesting complex II). Lane 1 shows an ideal banding pattern resulting from chloroplast isolation before solubilization and green gel electrophoresis on a 4-7% acrylamide gradient gel. Lanes 2 and 3 show average results from simple thylakoid isolation and electrophoresis on a non-gradient 5% acrylamide gel. Lanes 4-6 show increasing severity of under-solubilization. Lane 7 shows representative results for Arabidopsis using the simple protocol. Please click here to view a larger version of this figure.
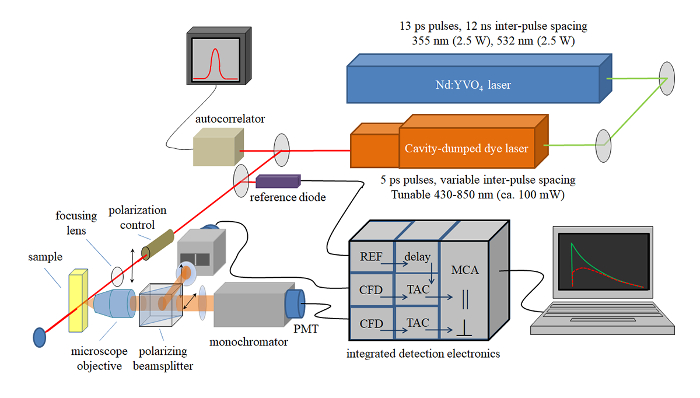
Figure 2: Schematic of time-correlated single photon counting instrument. Light source is a passively mode-locked NdYVO4 laser that pumps a cavity dumped dye laser. 5-ps pulses at variable repetition rates and wavelengths are characterized using an autocorrelator. The pulses are divided, with one portion going to a reference photodiode and the rest exciting the sample. Sample emission is collected using a microscope objective, and polarized components are detected using two detection channels. Sample emission is time resolved using the detection electronics indicated, where CFD = constant fraction discriminator, TAC = time-to-amplitude converter, and MCA = multi-channel analyzer. Time resolution is determined from the instrument response function (ca. 40 ps). Please click here to view a larger version of this figure.
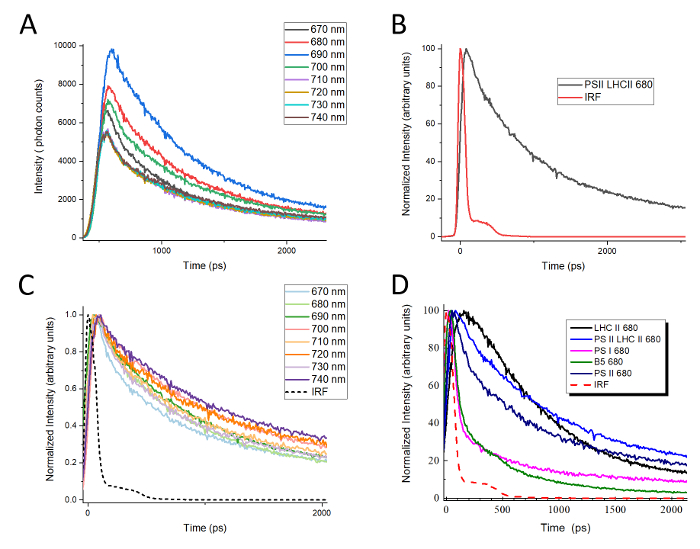
Figure 3: Representative raw TCSPC data and normalized fluorescence decay curves. (A) Overlay of raw TCSPC data for LHCII. TCSPC data was collected every 10 nm between 670 nm and 740 nm (B) A representative curve showing time registration of the fluorescence data to the instrument response function (IRF). (C) The LHCII data from (A) normalized to a maximum peak height of 1 after all curves were registered to their respective IRFs. (D) Overlay of fluorescence decay curves at 680 nm for PSI, PSII, PSII-LHCII, LHCII, and Band 5. All decay curves were normalized to a maximum peak height of 1. Please click here to view a larger version of this figure.
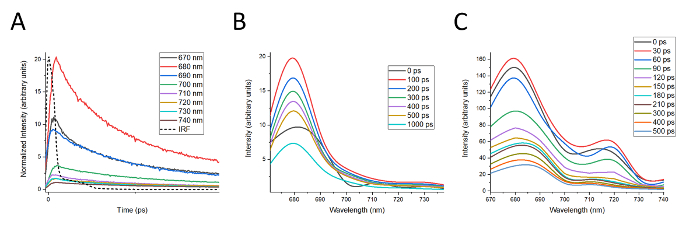
Figure 4: Construction of waterfall-style DAS. (A) Tail matching of the decay curves for LHCII in preparation for construction of DAS plots. (B) DAS plots for LHCII for time t = 0, for every 100 ps from 100 to 500 ps, and at 1000 ps. (C) DAS plots for Band 5 every 30 ps from t = 0 to 210 ps and every 100 ps thereafter to 500 ps. For both (B) and (C), time = 0 is defined by the IRF, which is 40 ps. Please click here to view a larger version of this figure.
