The whole procedure for in vitro expansion and metabolic labeling of primary embryonic NSPCs takes 6 days (Figure 1A). Quality of the BEND3 cell line and freshly isolated primary NSPCs are key to a successful experiment. BEND3 cells are the source of soluble factors that stimulate self-renewal and proliferation of NSPCs. It should be ensured that the BEND3 cells are free of any contamination and divide actively with minimal cell death before co-culturing with neural cells. The primary NSPCs must be carefully prepared to avoid excess damage during dissociation. Damaged NSPCs may still grow and differentiate; however, they are not able to respond to endothelial stimuli well to maintain stemness and expand. Extra caution should be taken to be aseptic during cell culturing, as the protocol does not suggest addition of antibiotics to the primary culture medium.
Successful endothelial co-culture will lead NSPCs to form large, sheet-like clones. Such featured clone shapes become evident at day 4 and are very typical at day 6. Within the clones, the cells maintain close contact with each other. Immunostaining with antibodies against the NSPC marker Nestin and the neuronal marker β-tubulin III should reveal that in the clone, most of the cells are Nestin+ NSPCs and very few are β-tubulin III+ neuronal cells. In contrast, the percentage of Nestin+ cells and β-tubulin III+ neuronal cells in clone formed in non-co-culture system are nearly the same (Figure 1B, 1D, and 1E).
The chemical reporter, Ac4ManNAz, is a metabolic analog and can be incorporated into the intrinsic protein sialylation pathway. High doses of Ac4ManNAz are toxic to cells. For each specific type of cell, the labeling concentration of Ac4ManNAz should be pre-tested to achieve the highest labeling efficiency without significant cytotoxicity. Here, the optimized labeling concentration of Ac4ManNAz for primary NSPC is 100 µM. Combinatory evaluation of cell death indicated by cellular and nuclei morphology suggests this labeling concentration does not cause obvious cytotoxic effects and is able to efficiently label NSPCs (Figure 1C and 1D). The clonal morphology, self-renewal, and differentiation potential of NSPCs in both the endothelial co-culture and non-co-culture system are not affected (Figure 1C, 1D, and 1E).
The successful labeling of NSPCs by Ac4ManNAz can be examined after conjugating biotin to a culture mediated by a bioorthogonal reaction between azide and alkyne. Every cell in the Ac4ManNAz-labeled culture is stained and visualized with Alexa Fluor 647-streptavidin. No cell is positive for Alexa Fluor 647-streptavidin staining in the DMSO control group. In addition, protein samples prepared from the Ac4ManNAz-labeled culture by biotin conjugation and streptavidin beads purification show strong Coomassie brilliant blue staining signal in SDS-PAGE gels. Meanwhile, there were only staining background and nonspecific binding signals in the lanes loaded with protein samples from the DMSO control group. This also indicates the efficient labeling of NSPCs by Ac4ManNAz (Figure 1F).
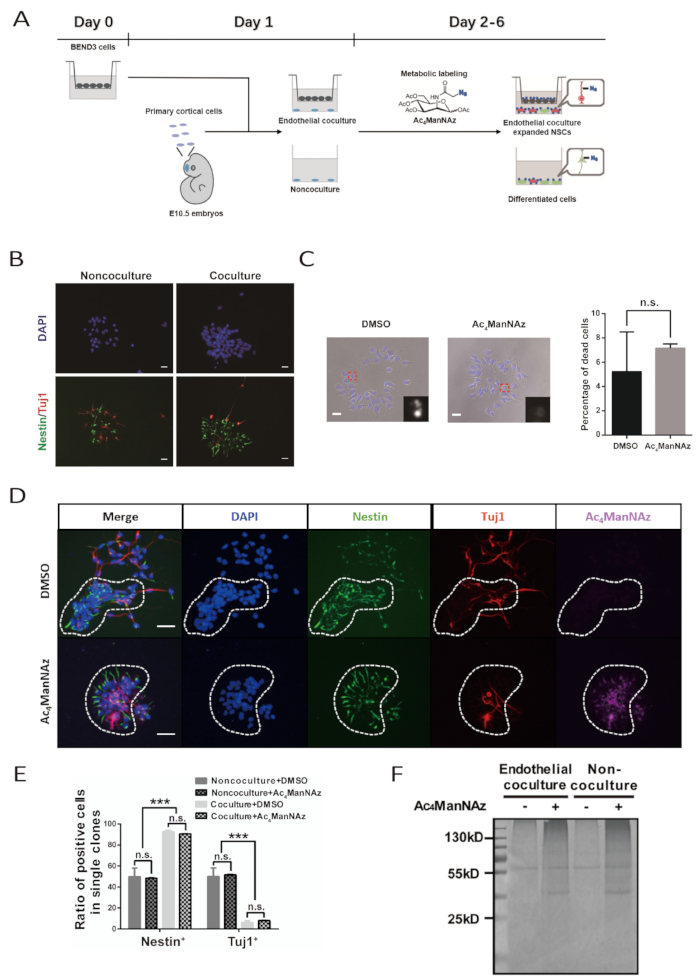
Figure 1: Identification of cell surface markers for primary NSPCs assisted by endothelial co-culture system and metabolic sialoglycan labeling. (A) Schematic of the workflow for the protocol. This figure has been modified from Bai et al.10. The BEND3 cells are seeded into matrix inserts on D0. The preparation of primary cortical NSPCs and set up of co-culture system are performed on D1. Metabolic labeling of culture lasts from D2 to D6. Culture refeeding is carried on D3 and D5. (B) The immunofluorescent images for clones formed by primary NSPCs after 5-day culture with or without endothelial cells. Scale bar indicates 20 µm. (C) Bright-field images for clones formed by primary NSPCs after a 5-day culture with Ac4ManNAz or DMSO. The nuclei were counterstained by DAPI. The scale bar indicates 20 µm. The error bar indicates SEM (n.s. = not significant). (D) The immunofluorescent images for NSPC formed clones in the endothelial co-culture with Ac4ManNAz or DMSO. Dashed circle demarcates a single neural clone. The scale bar indicates 50 µm. (E) Quantification of NSPCs and differentiated neurons in clones formed by NSPCs in endothelial co-culture and non-co-culture system with Ac4ManNAz labeling or DMSO control. The error bar indicates SEM (***p < 0.0005; n.s. = not significant). (F) Coomassie brilliant blue staining of proteins purified by streptavidin beads from neural cells labeled with Ac4ManNAz or DMSO in endothelial co-culture and nonco-culture system. The 55 kD band in control labelling groups represents nonspecific binding proteins. (B, C, E and F) corresponding to this protocol have been adapted from Bai et al.10. Please click here to view a larger version of this figure.
