Negli ultimi due decenni, solidi e film fatti di molecole organiche aggregate hanno trovato più applicazioni nei dispositivi optoelettronici. I dispositivi basati su tali materiali hanno molte proprietà interessanti, tra cui peso ridotto, flessibilità, basso consumo energetico e potenziale di produzione a basso costo utilizzando la stampa a getto d’inchiostro. I display basati su diodi a emissione di luce organica (OLED) stanno sostituendo i display cristallini liquidi come allo stato dell’arte per telefoni cellulari, computer portatili, televisori e altri dispositivi elettronici1,2,3,4. L’importanza degli OLED per le applicazioni di illuminazione dovrebbe aumentare nei prossimi anni4. Le prestazioni dei dispositivi fotovoltaici organici sono in costante miglioramento, con efficienze di conversione dell’energia superiori al 16% segnalate di recente per le celle solari organiche a singola giunzione5. I materiali organici hanno anche il potenziale di interrompere altre tecnologie, come le comunicazioni in fibra ottica, dove il loro utilizzo consente lo sviluppo di modulatori elettro-ottici con larghezze di banda estremamente elevate di 15 THz e oltre6,7.
Una sfida importante nell’ottimizzazione dei materiali molecolari allo stato solido per le applicazioni nell’optoelettronica è che in genere le loro proprietà dipendono fortemente dalla struttura su nanoscala del materiale. Il processo di produzione permette di definire la nanostruttura di un materiale in una certa misura utilizzando tecniche di crescita controllata, come la deposizione chimica del vapore,8 templating di molecole otticamente attive su un altro materiale (cioè una matrice polimerica9,10), annessione termica11,12,ecc. Tuttavia, il disturbo su nanoscala è intrinseco alla maggior parte dei materiali molecolari e di solito non può essere eliminato completamente. Pertanto, comprendere come il disordine influisce sulle proprietà di un materiale e trovare modi per progettarlo per prestazioni ottimali è essenziale per la progettazione razionale di materiali optoelettronici organici.
Il grado di disturbo nei materiali molecolari è di solito troppo grande per trattarlo come una perturbazione di una struttura cristallina periodica con una struttura elettronica che può essere descritta dalla teoria delle bande. D’altra parte, il numero di molecole che devono essere incluse in una simulazione per riprodurre le proprietà di un materiale sfuso o di un film è troppo grande per usare i primi principi metodi chimici quantistici come la teoria funzionale della densità (DFT)13,14 e la teoria funzionale della densità dipendente dal tempo (TD-DFT)15,16. Le molecole organiche con applicazioni nell’optoelettronica hanno in genere sistemi relativamente grandi, coniugati a z. molti hanno anche gruppi di donatori e accettanti. Catturare il corretto comportamento di trasferimento di carica in tali molecole è essenziale per calcolare le loro proprietà optoelettroniche, ma può essere realizzato solo utilizzando funzioni ibride corrette a lungo raggio in TD-DFT17,18,19,20. I calcoli che utilizzano tali funzioni scalano in modo super lineare con le dimensioni del sistema e, al momento, sono pratici solo per modellare le proprietà optoelettroniche di singole molecole organiche o piccoli aggregati molecolari che possono essere descritti utilizzando non più di 104 funzioni di base atomiche. Un metodo di simulazione che potrebbe descrivere materiali disordinati che consistono in un gran numero di cromofori sarebbe molto utile per la modellazione di questi sistemi.
La grandezza delle interazioni intermolecolari nei materiali molecolari è spesso paragonabile o inferiore all’ordine di variazione dei parametri energetici (come le energie eigenstate o le energie di eccitazione) tra le singole molecole che compongono il materiale. In questi casi, la modellazione multiscala è l’approccio più promettente per comprendere e ottimizzare le proprietà optoelettroniche di grandi sistemi molecolari disordinati21,22,23. Questo approccio utilizza metodi chimici quantistici di primo principi (di solito DFT e TD-DFT) per calcolare con precisione le proprietà delle singole molecole che compongono il materiale. L’Hamiltoniano di un campione di materiale abbastanza grande da rappresentare il materiale molecolare sfuso (forse, impiegando condizioni limite periodiche) viene quindi costruito utilizzando i parametri calcolati per le singole molecole. Questo hamiltoniano può quindi essere utilizzato per calcolare i parametri optoelettronici di un grande aggregato molecolare, una pellicola sottile o un materiale molecolare sfuso.
I modelli Exciton sono una sottoclasse di modelli multiscala in cui gli stati eccitati di un materiale molecolare sono rappresentati in una base di eccitoni: coppie elettrone-foro che sono legate dall’attrazione di Coulomb24,25. Per la modellazione di molti processi di stato eccitato, è sufficiente includere solo gli eccitoni Frenkel26, dove l’elettrone e il foro sono localizzati sulla stessa molecola. Gli eccitoni di trasferimento di carica, in cui l’elettrone e il foro sono localizzati su molecole diverse, potrebbero dover essere inclusi in alcuni casi (ad esempio, quando si modella la separazione della carica nei sistemi donatore-accettazione)27,28. Anche se i modelli exciton sono modelli multiscala che possono essere parametrizzati utilizzando solo calcoli di primo principio su singole molecole, tengono ancora conto delle interazioni intermolecolari. I due tipi di interazione primaria che possono rappresentare sono (a) accoppiamenti eccitonici tra molecole che caratterizzano la capacità degli eccitoni di delocalizzare o trasferire tra molecole e (b) la polarizzazione elettrostatica della densità elettronica su una molecola mediante la distribuzione di carica sulle molecole circostanti. Abbiamo dimostrato in precedenza che entrambi questi fattori sono importanti per la modellazione delle proprietà ottiche ed elettro-ottiche degli aggregati molecolari, come gli spettri di assorbimento ottico29 e le prime iperpolarizzabilità30.
In questo articolo, presentiamo un protocollo per la parametrizzazione di modelli di eccitamenti che possono essere utilizzati per calcolare gli spettri ottici e altre proprietà optoelettroniche di grandi aggregati molecolari e materiali molecolari sfusi. Si presume che l’hamiltoniano eccitonico sia un Hamiltoniano a derisioni24,,25,
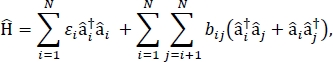
dove i è l’energia di eccitazione della molecola ith nel materiale, bij è l’accoppiamento eccitonico tra le molecole ith e jth, – i– e – i sono rispettivamente gli operatori di creazione e annientamento, per uno stato eccitato sulla molecola ith nel materiale. I parametri hamiltoniani eccitoni si trovano utilizzando calcoli TD-DFT che vengono eseguiti su singole molecole che compongono il materiale. In questi calcoli TD-DFT, la distribuzione della carica su tutte le altre molecole nel materiale è rappresentata dall’incorporazione elettrostatica di cariche a punti atomici per tenere conto della polarizzazione elettrostatica della densità elettronica di una molecola. Le energie di eccitazione, ii, per le singole molecole sono prese direttamente dall’uscita di calcolo TD-DFT. Gli accoppiamenti eccitonici, bij, tra le molecole vengono calcolati utilizzando il metodo cubo di densità di transizione31, con le densità di transizione da terra a eccitoperle per le molecole interagenti prelevate dall’output di un calcolo TD-DFT in Gaussian32 e post-elaborate utilizzando l’analizzatore d’onda multicompatibile 33. Per simulare le proprietà dei solidi molecolari sfusi, all’Hamiltoniano possono essere applicate condizioni limite periodiche.
Il protocollo corrente richiede che l’utente abbia accesso ai programmi Gaussian32 e Multiwfn33. Il protocollo è stato testato utilizzando Gaussian 16, revisione B1 e Multiwfn versione 3.3.8, ma dovrebbe funzionare anche per altre versioni recenti di questi programmi. Inoltre, il protocollo utilizza un’utilità personalizzata di C e un certo numero di script python 2.7 e Bash personalizzati, il cui codice sorgente viene fornito sotto la licenza generale GNU (versione 3) a https://github.com/kocherzhenko/ExcitonicHamiltonian. I calcoli devono essere eseguiti su una macchina che esegue un sistema operativo della famiglia Unix/Linux.
