Nas últimas duas décadas, sólidos e filmes feitos a partir de moléculas orgânicas agregadas encontraram múltiplas aplicações em dispositivos optoeletrônicos. Dispositivos baseados em tais materiais têm muitas propriedades atraentes, incluindo pequeno peso, flexibilidade, baixo consumo de energia e potencial para produção barata usando impressão a jato de tinta. Displays baseados em diodos emissores de luz orgânica (OLEDs) estão substituindo displays cristalinos líquidos como estado da arte para telefones celulares, laptops, televisores e outros dispositivos eletrônicos1,2,3,4. Espera-se que a importância dos OLEDs para aplicações de iluminação aumente nos próximos anos4. O desempenho dos dispositivos fotovoltaicos orgânicos está melhorando continuamente, com eficiências de conversão de energia acima de 16% recentemente relatadas para células solares orgânicas de junção única5. Os materiais orgânicos também têm o potencial de interromper outras tecnologias, como as comunicações de fibra óptica, onde seu uso permite o desenvolvimento de moduladores eletro-ópticos com larguras de banda extremamente altas de 15 THz e acima de6,7.
Um grande desafio na otimização de materiais moleculares de estado sólido para aplicações em optoeletrônica é que normalmente suas propriedades dependem fortemente da estrutura nanoescala do material. O processo de produção permite definir a nanoestrutura de um material em certa medida usando técnicas de crescimento controlado, como deposição de vapor químico,8 templating de moléculas opticamente ativas em outro material (ou seja, uma matriz de polímeros9,10),recozimento térmico11,12, etc. No entanto, a desordem nanoescala é intrínseca à maioria dos materiais moleculares e geralmente não pode ser totalmente eliminada. Portanto, entender como a desordem afeta as propriedades de um material e encontrar formas de projetá-lo para um desempenho ideal é essencial para o design racional de materiais optoeletrônicos orgânicos.
O grau de desordem em materiais moleculares é geralmente muito grande para tratá-lo como uma perturbação de uma estrutura cristalina periódica com uma estrutura eletrônica que pode ser descrita pela teoria da banda. Por outro lado, o número de moléculas que devem ser incluídas em uma simulação para reproduzir as propriedades de um material a granel ou de um filme é muito grande para usar os primeiros princípios de métodos químicos quânticos como a teoria funcional da densidade (DFT)13,,14 e a teoria funcional da densidade dependente do tempo (TD-DFT)15,16. Moléculas orgânicas com aplicações em optoeletrônica tipicamente têm sistemas π-conjugados relativamente grandes; muitos também têm grupos doadores e aceitadores. Capturar o comportamento correto de transferência de carga em tais moléculas é essencial para calcular suas propriedades optoeletrônicas, mas só pode ser realizado usando funcionais híbridos corrigidos de longo alcance em TD-DFT17,,18,19,20. Cálculos que usam tais funções escalam super linearmente com o tamanho do sistema e, atualmente, são apenas práticos para modelar as propriedades optoeletrônicas de moléculas orgânicas individuais ou pequenos agregados moleculares que podem ser descritos usando não mais do que ~104 funções de base atômica. Um método de simulação que poderia descrever materiais desordenados que consistem em um grande número de cromóforos seria muito útil para modelar esses sistemas.
A magnitude das interações intermoleculares em materiais moleculares é muitas vezes comparável ou menor do que a ordem de variação nos parâmetros energéticos (como as energias eigenstate ou energias de excitação) entre moléculas individuais que compõem o material. Nesses casos, a modelagem multiescala é a abordagem mais promissora para compreender e otimizar as propriedades optoeletrônicas de grandes sistemas moleculares desordenados21,22,23. Esta abordagem usa métodos químicos quânticos de primeiros princípios (geralmente DFT e TD-DFT) para calcular com precisão as propriedades das moléculas individuais que compõem o material. O hamiltoniano de uma amostra de material que é grande o suficiente para representar o material molecular a granel (talvez, empregando condições periódicas de limite) é então construído usando os parâmetros que foram calculados para moléculas individuais. Este hamiltoniano pode então ser usado para calcular os parâmetros optoeletrônicos de um grande agregado molecular, um filme fino, ou um material molecular a granel.
Os modelos exciton são uma subclasse de modelos multiescala em que estados animados de um material molecular são representados em uma base de excitons: pares de buracos de elétrons que são vinculados pela atração de Coulomb24,25. Para modelar muitos processos de estado animados, basta incluir apenas os excitons frenkel26, onde o elétron e o buraco são localizados na mesma molécula. Os excitons de transferência de carga, onde o elétron e o orifício são localizados em diferentes moléculas, podem precisar ser incluídos em alguns casos (por exemplo, ao modelar a separação da carga em sistemas de aceitação de doadores)27,28. Embora os modelos de exciton sejam modelos multiescala que podem ser parametrizados usando apenas cálculos de primeiro princípio em moléculas individuais, eles ainda explicam interações intermoleculares. Os dois tipos de interação primária que eles podem explicar são (a) acoplamentos excitonicos entre moléculas que caracterizam a capacidade de excitons de deslocalizar ou transferir entre moléculas e (b) polarização eletrostática da densidade eletrônica em uma molécula pela distribuição de carga em moléculas circundantes. Já mostramos anteriormente que ambos os fatores são importantes para modelar as propriedades ópticas e eletro-ópticas dos agregados moleculares, como o espectro de absorção óptica29 e as primeiras hiperpolarizações30.
Neste artigo, apresentamos um protocolo para parametrização de modelos de excitoque que podem ser usados para calcular os espectros ópticos e outras propriedades optoeletrônicas de grandes agregados moleculares e materiais moleculares a granel. O hamiltoniano excitonico é assumido como um hamiltoniano apertado24,25,
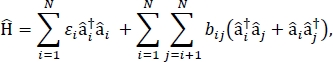
onde εi é a energia de excitação da iªth molécula no material, bij é o acoplamento excitonico entre as moléculas ith e jth, â i† e â i são os operadores de criação e aniquilação, respectivamente, para um estado animado na molécula ith no material. Os parâmetros hamiltonianos excitonicos são encontrados usando cálculos TD-DFT que são realizados em moléculas individuais que compõem o material. Nestes cálculos TD-DFT, a distribuição de carga em todas as outras moléculas do material é representada pela incorporação eletrostática de cargas de ponto atômico para explicar a polarização eletrostática da densidade eletrônica de uma molécula. As energias de excitação, εi, para moléculas individuais são retiradas diretamente da saída de cálculo TD-DFT. Os acoplamentos excitonicos, bij, entre moléculas são calculados usando o método cubo de densidade de transição31,com as densidades de transição de estado terra-para-excitação para as moléculas interativas retiradas da saída de um cálculo TD-DFT em Gaussian32 e pós-processados usando o analisador multifuncional multifuncional de função de onda33. Multiwfn Para simular as propriedades dos sólidos moleculares a granel, condições periódicas de limite podem ser aplicadas ao hamiltoniano.
O protocolo atual exige que o usuário tenha acesso aos programas Gaussian32 e Multiwfn33. O protocolo foi testado usando gaussian 16, revisão B1 e Multiwfn versão 3.3.8, mas também deve funcionar para outras versões recentes desses programas. Além disso, o protocolo usa um utilitário C++ personalizado e uma série de scripts personalizados python 2.7 e Bash, o código-fonte para o qual é fornecido sob a Licença Pública Geral GNU (Versão 3) em https://github.com/kocherzhenko/ExcitonicHamiltonian. Os cálculos devem ser realizados em uma máquina executando um sistema operacional da família Unix/Linux.
