rbm-3.2 is a putative RNA-binding protein that has homology to human cleavage stimulation factor subunit 2 tau variant (WormBase: https://wormbase.org/species/c_elegans/gene/WBGene00011156#0-9fcb6d-10). The RBM-3.2 protein was identified as a binding partner of Protein Phosphatase 1 (GSP-1) and its regulators Inhibitor-2 (I-2SZY-2) and SDS-22 in a previous study that identified these proteins as novel regulators of C. elegans centriole duplication (data not shown)45. Presently, very little is known regarding the function of the rbm-3.2 gene in C. elegans. Hence, to further investigate the biological role of the C. elegans rbm-3.2 gene, the rbm-3.2-null allele rbm-3.2(ok688) was obtained from the Caenorhabditis Genetics Center (CGC).
Unfortunately, in addition to possessing a deletion of the entire rbm-3.2 gene, the rbm-3.2(ok688) allele also results in a partial deletion of an overlapping gene, rbm-3.1, thereby complicating genetic analysis. Therefore, in order to accurately investigate the role of the rbm-3.2 gene in C. elegans, we used CRISPR/Cas9 editing to introduce three premature stop codons very close to the start of the C. elegans rbm-3.2 coding region, leaving the overlapping rbm-3.1 gene intact.
To introduce these premature stop codons into the C. elegans rbm-3.2 gene, we designed a crRNA with a PAM motif that was located on the opposite strand (template strand) of DNA (Figure 1A). The Cas9 cut site was located 6 bases away from the rbm-3.2 start codon ATG. To introduce premature stop codons into the rbm-3.2 gene, we designed a repair template with the following five major characteristics: 1) 35 bases of uninterrupted homology to rbm-3.2 upstream of the rbm-3.2 start codon (left homology arm) 2) A short stretch of bases containing the PAM motif was deleted 3) An EcoRI restriction site was introduced immediately after the start codon for screening 4) The second and the third codons of rbm-3.2 were deleted and three stop codons were introduced after the fifth RBM-3.2 codon to stop translation of the rbm-3.2 mRNA 5) 35 bases of uninterrupted homology to the first intron of rbm-3.2 were included downstream of the edit (right homology arm) (Figure 1B).
The injection mix for this CRISPR experiment was prepared as indicated in Table I. The injection mix was incubated at 37 °C for 15 minutes to assemble ribonucleoprotein complexes. The mix was then centrifuged and loaded into the pulled microinjection needle. Microinjection into the C. elegans gonad was performed as described in Iyer et al. 201943.
Figure 2 depicts the experimental timeline for generating edited C. elegans using this protocol. Although we typically inject 30 worms for each CRISPR experiment, some laboratories inject fewer worms (between 10 to 20) for each CRISPR experiment. Since injecting more worms increases the probability of finding positive edits, we prefer to inject a larger number of worms. Many laboratories pick "jackpot" broods (plates with greater than 50% Rol and Dpy progeny) for screening. In our experience after performing several CRISPR experiments, although jackpot broods do arise occasionally, a majority of times, the Rol worms that are picked for screening often come from many different P0 plates, each consisting of a few Rol progeny. No jackpot broods were obtained in this experiment.
In total, we screened the genomic DNA of 73 F1 Rol worms that were obtained from 7 injected P0 worms for the presence of the edit. The sequences of the screening primers and their locations with respect to the start codon of the rbm-3.2 gene are represented in Figure 3A. Through our analysis, 7 out of 73 F1 worms were found to be positive for the edit (9.5%) (Figure 3B). Unedited worms displayed a single DNA fragment of 445 bp upon EcoRI digestion. Whereas, worms carrying a premature stop codon in the rbm-3.2 gene exhibited two fragments of 265 bp and 166 bp respectively upon EcoRI digestion. Heterozygous-edited worms displayed three fragments upon EcoRI digestion: one wild-type unedited DNA fragment of 445 bp and two DNA fragments of 265 bp and 166 bp respectively from the edited copy of the rbm-3.2 gene.
To identify worms carrying homozygous edits, we transferred 12 F2 worms from the identified positive F1 heterozygotes onto new individual plates and allowed them to produce progeny (F3). The F2 worms were then screened for homozygosity as described earlier. 6 out of 12 (50%) screened worms were found to be homozygous for our edit of interest (Figure 4A). The genomic DNA of an identified homozygous-edited worm line was used to set up a DNA sequencing reaction. DNA sequencing analysis confirmed the presence of the edit in its homozygous state (Figure 4B). In general, it is beneficial to sequence multiple homozygous worm lines to ensure that all homozygous-edited worm lines exhibit the same phenotype (if the null-mutation produces a specific phenotype). Further, it might also be necessary to validate gene knockouts using an expression-based assay such as western blotting or RT-PCR or via phenotype analysis. This is because, it is possible that in-spite of introducing premature stop codons at the beginning of the gene, an alternative ATG might be present downstream of the premature stop codons. This ATG could be used to initiate protein expression, resulting in a partially-functional truncated protein.
| Reagent | Concentration | Volume to Add |
| Cas9 protein in nuclease-free water with 20% glycerol | 2 µg/µL | 5 µL |
| tracrRNA in 5 mM Tris-Cl pH 7.5 | 4 µg/µL | 5 µL |
| dpy-10 crRNA in 5 mM Tris-Cl pH 7.5 | 8 µg/µL | 0.4 µL |
| rbm-3.2 crRNA in 5 mM Tris-Cl pH 7.5 | 8 µg/µL | 1 µL |
| dpy-10 repair template in sterile nuclease-free water | 500 ng/µL | 0.55 µL |
| rbm-3.2 repair template in sterile nuclease-free water | 1 µg/µL | 2.2 µL |
| KCl in sterile nuclease-free water | 1 M | 0.5 µL |
| sterile nuclease-free water | – | 5.35 µL |
Table 1: Components of the injection mix for CRISPR/Cas9 editing using ribonucleoprotein complexes and dpy-10 as a co-CRISPR marker. Use sterile techniques and RNase-free reagents while making the injection mix. Please note that sterile, non-DEPC treated nuclease-free water was used to make the injection mix.
| Reagent | Concentration | Volume to add |
| KCl | 1 M | 5 mL |
| Tris-HCl pH 8.3 | 1 M | 1 mL |
| MgCl2 | 1 M | 250 µL |
| NP-40 (or IGEPAL CA-630) | 100% | 450 µL |
| Tween-20 | 100% | 450 µL |
| Gelatin | 2% | 500 µL |
| Water | – | 92.35 mL |
Table 2: Worm lysis buffer recipe. The worm lysis buffer can be made in bulk, autoclaved, filtered and aliquoted for long term storage (NOTE: the lysis buffer can be stored for over a year at room temperature). Add a 1:100 dilution of 20 mg/mL proteinase K to the worm lysis buffer just before each use.
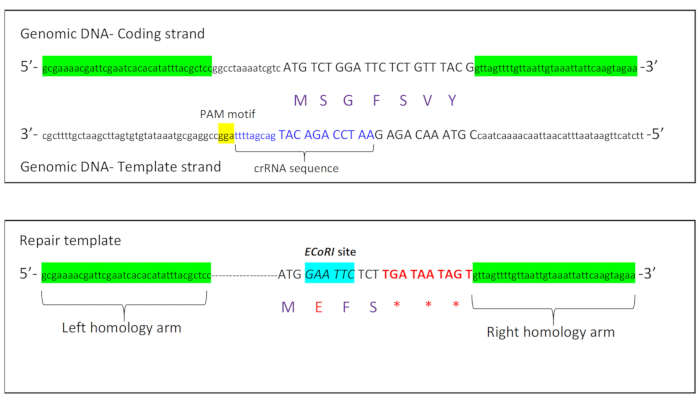
Figure 1: Schematic showing crRNA and repair template design for rbm-3.2 premature stop CRISPR. A. Schematic displaying the coding and template strands of the rbm-3.2 gene with the crRNA sequence and the PAM motif sequence located on the template strand. B. Schematic representing the different characteristics of the repair template that was synthesized to introduce three premature stop codons into the rbm-3.2 gene. Please click here to view a larger version of this figure.

Figure 2: Experimental timeline for generating homozygous-edited C. elegans by CRISPR/Cas9 editing using preassembled ribonucleoprotein complexes and dpy-10 as a co-CRISPR marker. A day-by-day breakdown of the steps that need to be performed to generate homozygous-edited C. elegans using this method of CRISPR/Cas9 editing. Briefly, 30 worms were injected with the CRISPR editing mix using microinjection and segregated onto individual MYOB plates seeded with OP50 E. coli. After 24 hours, the injected P0 worms were transferred onto fresh new MYOB plates and allowed to continue laying eggs. On Days 3 and 4 after microinjection, the plates were examined for the presence of Rol F1 worms. The plates with the maximum number of Rol and Dpy F1 worms were selected and 73 F1 Rol worms (we usually pick between 50 to 100 F1 Rol worms per CRISPR experiment) were singled onto new individual MYOB plates (1 worm per plate) and allowed to lay eggs for about 2 days. On Day 6 after microinjection, worm lysates were prepared from the F1 worms that had produced progeny (F2) and were screened for the presence of the edit by PCR followed by restriction digestion with EcoRI and agarose gel electrophoresis. On Day 7, 12 non-Rol, non-Dpy F2 worms were transferred from the positive plates onto new individual plates and allowed to produce progeny (F3). On Day 9, the F2 worms were screened for homozygosity of the edit as described previously. On Day 10, worm lysis, PCR and PCR cleanup were performed for the homozygous F3 worms and the DNA concentration was measured using a NanoDrop spectrophotometer. On Day 11, Sanger sequencing reactions were set up for positive samples and the reactions were sent for DNA sequencing. On Day 12, the sequencing results were analyzed, and the presence of the edit was verified using a sequence analysis software (e.g. CLC sequence viewer). Please click here to view a larger version of this figure.
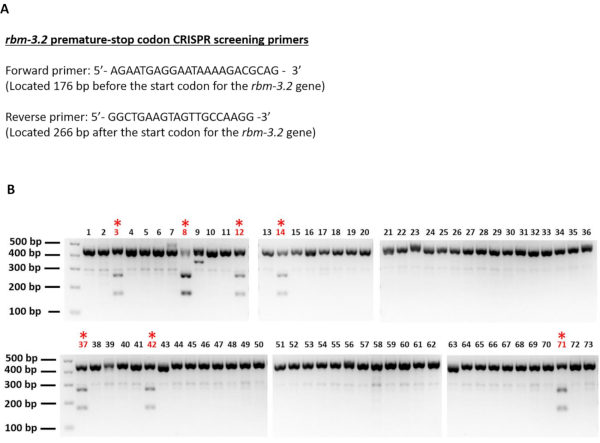
Figure 3: Screening for C. elegans that are heterozygous for the rbm-3.2 premature stop codons. Agarose gel electrophoresis images of C. elegans genomic PCR DNA digested with EcoRI. 73 individual F1 worms were genotyped and screened for the presence of the rbm-3.2 edit. Red numbers and asterisks indicate positively-edited worms. All the 7 identified positively-edited C. elegans were heterozygous for the edit as they exhibited one wild-type unedited DNA fragment of 445 bp and two DNA fragments of 265 bp and 166 bp respectively from the edited copy of the rbm-3.2 gene upon EcoRI digestion. Please click here to view a larger version of this figure.
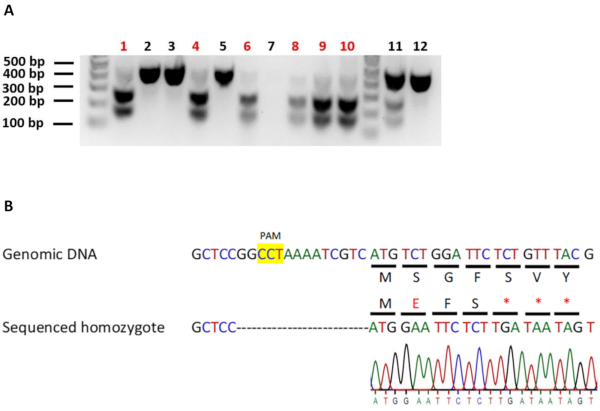
Figure 4: Identifying and verifying homozygous-edited C. elegans carrying the rbm-3.2 premature stop codons. A. Screening for C. elegans that are homozygous for the rbm-3.2 premature stop codons. Agarose gel electrophoresis images of C. elegans genomic DNA digested with EcoRI. 12 individual F2 worms from positive plates were genotyped and screened for homozygosity of the rbm-3.2 edit. Red: homozygous-edited worms. 6 out of the 12 screened F2 worms (50%) were homozygous for the rbm-3.2 premature stop codons. No PCR product was present for worm 7. B. Confirming the insertion of the premature stop codons in rbm-3.2 by DNA sequencing. Schematic showing the comparison of DNA and protein sequences of unedited and edited homozygotes. Analysis of DNA sequencing results of genomic DNA from homozygous-edited worms confirmed the presence of the three premature stop codons in the rbm-3.2 gene upon CRISPR/Cas9 editing. All the resultant amino acid changes after CRISPR/Cas9 editing are indicated in either red letters or red asterisks. Please click here to view a larger version of this figure.
