1. Cell plating
- Grow HFK LXSN and HFK 8E6 cells in 10 cm plates in keratinocyte culture media (10 mL/plate) with human keratinocyte growth supplement (HKGS) and 1% penicillin/streptomycin. Grow cells to about 80% confluence at 37 °C in a jacket incubator with 5% CO2.
- Replace culture media with 3 mL of trypsin-EDTA (0.05%, ethylenediamine tetraacetic acid). Incubate at 37 °C for 3 min. Neutralize trypsin with equal volume of fetal bovine serum (FBS) supplemented media and transfer cells to a 15 mL centrifuge tube. Centrifuge at 300 x g for 5 min.
- Resuspend cells with 10 mL of keratinocyte culture media with HKGS. Determine the concentration of cells with a hemocytometer.
- Plate 4 x 105 cells/6-cm plates (seed two plates for HFK LXSN and two plates for HFK 8E6) in 4mL of keratinocyte culture media with HKGS and 1% penicillin and streptomycin. Grow at 37 °C in a jacket incubator with 5% CO2.
NOTE: Analysis of HFK cells was chosen for this protocol for two reasons. First, HFK is a difficult to transfect cell line. Thus, by demonstrating that the protocol works in this cell line, evidence is provided that it will likely work in more commonly used and more readily transfected cells. Secondly, previously published data demonstrate that a viral protein (8E6) hinders the repair of double strand breaks in DNA20,21,22. Thus, comparing HFK LXSN and HFK 8E6 allows us to demonstrate the ability of the assay to detect increases in mutations associated with a reduction in cellular repair capacity.
2. Transfection
- On the day of transfection (24 h after plating), replace media with 3 mL of antibiotic-free supplemented media. Incubate for 2 h at 37 °C in a jacket incubator.
- Transfect cells with appropriate lipid-based transfection reagents according to the manufacturer's instructions.
- Warm transfection reagents to room temperature and pipette gently before using.
- For each cell line (HFK LXSN and HFK 8E6), place appropriate amount of transfection buffer (as directed by the manufacturer) in a sterile 1.5 mL centrifuge tube (Tube 1). Include another tube with the same amount of transfection buffer (mock transfection or Tube 2).
- Add 2 µg of plasmid DNA expressing CAS9/sgRNA targeting human CD4 (px330-CD4, 5'- GGCGTATCTGTGTGAGGACT) to Tube 1 from step 2.2.2. Pipette gently to mix completely. Add equal volume of sterile water to Tube 2.
- Include a control plate with transfection reagents alone (no plasmid) for each cell line.
NOTE: The second plate serves as a negative control in the experiment, allowing the user to confirm that transfection with the CAS9/sgRNA are not responsible for any mutations. - Add appropriate amount of the transfection reagent (as directed by the manufacturer) to the tube with DNA mixture (Tube 1) from step 2.2.3 and the mock transfection (Tube 2) from step 2.2.2. Pipette gently to mix completely. Incubate at room temperature for 15-30 min to allow sufficient time for complexes to form.
- Add the transfection mixture drop-wise to the plate. Gently rock the culture for 1 min to evenly distribute the transfection mixture.
- Incubate for 48 h after the transfection to allow CAS9 expression.
- Harvest cells by trypsinization.
- Replace culture media with 1 mL of trypsin-EDTA (0.05%, ethylenediamine tetraacetic acid). Incubate at 37 °C for 3 min. Neutralize trypsin with equal volume of FBS supplemented media.
- For each plate of cells, transfer the cell suspension to two microcentrifuge tubes with equal aliquots. Centrifuge at 300 x g for 5 min.
- Resuspend the cell pellet from one tube in step 2.4.2 in 1 mL of phosphate buffered saline (PBS) for sequencing. Resuspend the other tube from step 2.4.2 with ice-cold PBS for immunoblot.
- Harvest the whole cell lysates for immunoblot.
- Centrifuge the tube at 300 x g for 5 min. Discard the supernatant.
- Add 100 µL radioimmunoprecipitation assay buffer (RIPA lysis buffer) mixed with 1 % protease inhibitor and 1% phosphatase inhibitor into the tube, mix thoroughly with a pipette and incubate for 10 min on ice.
NOTE: RIPA lysis buffer contains 10 mM Tris-HCl, pH 8.0; 1 mM EDTA; 0.5 mM EGTA; 1% Triton X-100; 0.1% Sodium Deoxycholate; 0.1% SDS; 140 mM NaCl, and deionized water. - Centrifuge lysates at 13,000 x g for 10 min. Collect supernatants for immunoblot.
3. Measuring CAS9 expression via immunoblot
- Determine the protein concentration with a bicinchoninic acid (BCA) assay according to the manufacturer's instructions.
- Run 20 µg of protein of each sample in the wells of a 3%-8% Tris-acetate gel for 150 min and semidry transfer (10 V for 30 min and then 25 V for 12 min) to polyvinylidene difluoride membrane.
- After blocking the membrane in 5% nonfat dry milk in PBS with 0.1% tween (PBST) for 1 h at room temperature, add anti-CAS9 (1:1000) and anti-GAPDH (1:1000) antibodies. Incubate at 4 °C overnight.
- After washing the membrane with PBST, incubate the membrane with secondary antibody in 5% nonfat dry milk in PBST for 1 h at room temperature.
- Image the blot and determine CAS9 level by densitometry23. See Figure 1 for a representative blot.
NOTE: Detecting phosphorylated H2AX (S139) foci formation by immunofluorescence microscopy can be used to validate CAS9 activity14. A low number of distinct foci (typically 1-4 foci) are expected depending on the cell cycle position, whether mutations in the CAS9 target site prevent further cutting, and how many copies of the CAS9 cutting site exist in the genome of interest. A representative image is shown in Figure 2.
4. Nucleic acid extraction and amplicon generation
- Extract DNA from cell samples from step 2.4.3 using a high-molecular weight DNA extraction kit, as specified by the manufacturer.
- Resuspend primers with indicated solvent according to the datasheet. Dilute with the same reagent to 20 µM and pool 20 µM primers into the indicated pools.
NOTE: Primer pool is listed in Supplemental Table 1. - Create a PCR Master mix using a long amplification Taq polymerase for each 20 µM primer pool as specified in Table 1.
- Add 21 µL of the mastermixes to separate PCR tubes.
- Add 4 µL of the target sample (100 ng/µL) from step 4.1 to PCR assay tubes containing mastermix and cap assay tubes. Ensure separate reactions for each primer pool.
- Vortex to mix PCR assay tubes and centrifuge (quick spin) to remove droplets from tube lids.
- Place the PCR tubes on a conventional thermal cycler machine.
- Program PCR machine as specified in Table 2.
- Run the program on a thermal cycler.
5. PCR clean-up
- Remove primers from PCR reactions using a bead-based PCR cleanup system.
- Remove clean-up beads from the refrigerator 30 min prior to use.
- Vortex beads well prior to use and ensure all beads are resuspended.
- Add 30 µL (1.2x) of resuspended beads to each well of a deep well 96-well plate.
- Add 25 µL of PCR reaction to wells containing beads.
- Place the plate on a plate shaker at 2000 rpm for 2 min.
- Allow the plate to remain at room temperature for 5 min following shaking.
- Place the deep well plate on a 96-well plate magnet and incubate for 2 min.
- Remove and discard the supernatant without the disturbing beads.
- While the plate remains on the magnet, add 180 µL of 80% ethanol and incubate for 30 s. Remove and discard the supernatant.
- Repeat step 5.1.9.
- Using a 10 µL pipette, remove and discard any remaining liquid from the wells.
- Allow beads to dry at room temperature for 10 min.
- Add 20 µL of nuclease free water to the wells containing beads and remove the plate from the magnet.
- Shake the plate at 2000 rpm for 2 min at room temperature.
- Incubate the plate at room temperature for 5 min.
- Place the plate on a magnet stand and incubate for 2 min at room temperature.
- Remove the supernatant into a second, labeled PCR plate. This contains the cleaned-up DNA.
- Measure the concentration of each reaction with a Fluorometer.
- Ensure that dsDNA Fluorometer reagents are at room temperature.
- Set up Fluorometric assay tubes plus two additional tubes for standards.
- Add 199 µL of 1x dsDNA working solution to all but two tubes. Add 190 µL of the working solution to the last two tubes.
- Add 10 µL of the two standards (included in the Table of Materials) to separate the assay tubes.
- Add 1 µL of each PCR reaction to Fluorometer mastermix tubes.
- Vortex tubes to mix and incubate at room temperature for 2 min.
- On the home screen of the fluorometer, select the button with the assay kitin use (1x dsDNA) then select Read Standards and Run Samples.
- Insert standard 1 tube, select the Read button and then repeat for standard 2.
- Following step 5.2.6, repeat for one sample, select a sample volume of 1 µL and the resulting concentration will be provided.
- Repeat step 5.2.7 for the remaining samples.
- Calculate the projected molarity of all reactions and pool equal concentrations of reactions from each individual sample separately (one final pool per sample) using the equation below.
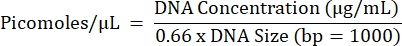
- Repeat steps 5.2.1 to 5.2.7 to obtain the final pool concentration.
- Check the amplicon pool on a capillary electrophoresis machine/agarose gel as specified by the manufacturer.
- Prepare capillary electrophoresis tubes for the appropriate number of samples. Add 7 µL of the DNA buffer as specified by the manufacturer.
- Add 4 µL of the amplicon pool to the tube containing DNA buffer.
- Place tubes on the electrophoresis machine and run the machine as specified by the manufacturer for dsDNA.
- View the electrophoresis gel pictures ensuring the bands localize to ~5 kb (size of amplicons).
- Calculate the projected molarity of all reactions and pool equal concentrations of reactions from each individual sample separately (one final pool per sample) using the equation below.
6. Library preparation
- Dilute sample pools from step 5.3.1 to 0.2 ng/µL for library preparation.
- Using a low-input library preparation kit compatible with short sequences (300bp) prepare libraries using unique index combinations for each sample pool created in step 5.3 following the manufacturer's instructions.
NOTE: Follow the manufacturer's instructions to select index sequences. All indexes amenable to the library prep kit will work for the samples. - Following library preparation, pool all samples according to the manufacturer's instructions.
NOTE: Before creating the library pool, calculate the number of reads necessary for 250x coverage of your target sequence and ensure that the selected sequencing cartridge can provide adequate coverage for each included sample. For 0.5Mb total, this will equate to 1M reads.
- Using a low-input library preparation kit compatible with short sequences (300bp) prepare libraries using unique index combinations for each sample pool created in step 5.3 following the manufacturer's instructions.
- Prepare the library pool for sequencing.
- Thaw and prepare a 300-cycle cartridge and sequencing reagents.
- Denature and dilute the sequencing pool created in step 5.1.1 according to the sequencer's manufacturer's instructions.
- Add denatured and diluted library pool to sequencing reagents and Run the sequencing machine as specified by the manufacturer.
NOTE: See attached Table 3 for trouble shootings.
7. Data analysis
NOTE: All data steps are performed in the genomic data analysis software. Parentheses indicate user input. Greater than sign indicates the order of mouse clicks for any given step (e.g., 1st mouse click>2nd mouse click)
- Import the reads by clicking on Open Software > Import > Illumina > Select Files > Next > Select location to Save > Finish. The reads will now appear in the software.
- Trim and filter the reads.
- In the deep sequence data analysis software, trim the raw reads default parameters.
- Highlight the reads and click Toolbox > Prepare Sequencing Data > Trim Reads > Next > Next > Next > Next > Save > Next > (Select location to save) > Finish
- Map the trimmed reads to reference.
- Map trimmed reads to the reference sequence used in step 4.1 using a match score of 2, mismatch cost of 3 and insertion/deletion costs of 2. Ensure that the length fraction is above 0.7 and similarity fraction is at or above 0.8.
- Highlight the trimmed read file and click Toolbox > Resequencing Analysis > Map Reads to Reference > Next > (Select reference sequence) > Next > (Ensure parameters are indicated as above) > Next > Save > (Select location to save) > Finish.
- Extract variants and indels.
- Using an appropriate indel caller, extract indels using a p-value threshold of 0.005 or lower and a maximum number of mismatches of 3.
- Highlight mapped read file and click Toolbox > Resequencing Analysis > Variant detection > Indels and Structural Variants > Next > (Ensure required significance is input > Next > Save > (Select location to save) > Finish.
- Using an appropriate variant caller, call variants from the read mapping using a significance of 5%.
- Highlight mapped read file and click Toolbox > Resequencing Analysis > Variant Detection > Low frequency variant detection > Next > (Ensure required significance is input > Next > Next > Save > (Select location to save) > Finish.
NOTE: Ensure to account for the ploidy of the host genome in the indel and variant callers. Do not extract mutants below 5%. This threshold accounts for PCR and sequencing errors associated with the assay. Normalization (based on immunoblot detection of CAS9) should be done by adjusting sequencing coverage. For example, if sample A has twice the transfection efficiency of sample B, then 50% of the reads from sample A should be used for analysis. This should be done by random sampling and not reduce the coverage for any sample below 100x.
Three representative results are presented for this protocol. Figure 1 is an immunoblot confirming expression of CAS9 in HFK control (LXSN) and HFK expressing beta-HPV 8E6 (8E6). 48 h after transfection, whole cell lysates were harvested and subsequently probed with an anti-CAS9 antibody (or GAPDH as a loading control). The result shows that HFK LXSN and HFK 8E6 are expressing similar amount of CAS9 indicating that transfection efficiency is similar between two cell lines. Figure 2 is an immunofluorescence microscopy image showing CAS9 induced DSBs using pH2AX foci, a standard marker for DSBs24. This indicates that two DSBs are induced by CAS9/sgRNA and thus confirms that DSB induction is occurring as expected. Figure 3 shows that 8E6 increases genomic variations within 200 Kb around the CAS9 induced DSB22. This is consistent with the hypothesis that HPV8 E6 deregulates DSB repair and increases genomic instability. This figure shows one way in which data obtained from this approach can be displayed.
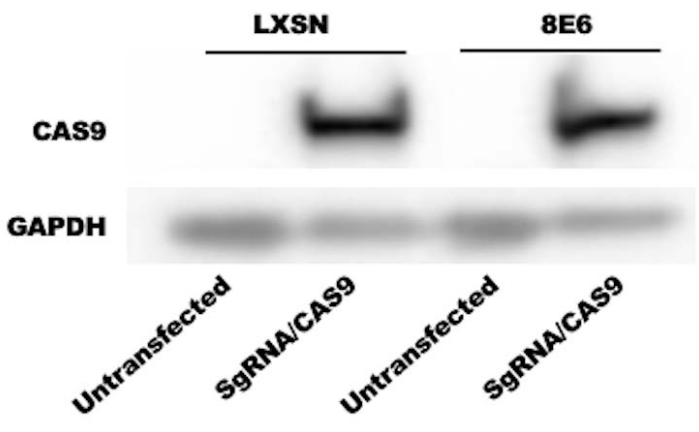
Figure 1: Representative image of immunoblot comparing CAS9 expression in untransfected and transfected HFK cells. sgRNA/CAS9 plasmids were used to transfect HFK cells. Anti-CAS9 antibody was used to detect the CAS9 protein. GAPDH is used as a loading control. Please click here to view a larger version of this figure.
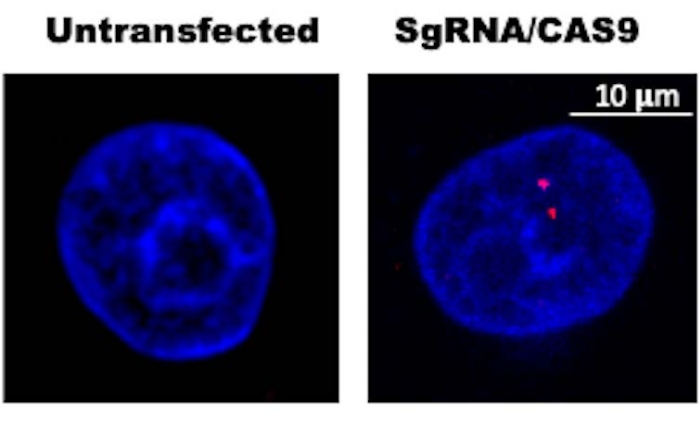
Figure 2: Representative immunofluorescence image of phosphorylated histone H2AX (S139) in sgRNA/CAS9 transfected cell and untransfected control. DAPI was used to stain DNA (Blue). pH2AX (Red) is a marker for CAS9-induced DSB. This demonstrates that optimal CAS9 expression has been achieved by the absence of off-target cleavage. Depending on the number of targeted genome loci (altered by changes in ploidy of the cell, target site copy number variations, or cell cycle position), the number of foci could be higher. However, any increase should be predictable based on the cell type and target site analyzed. Please click here to view a larger version of this figure.
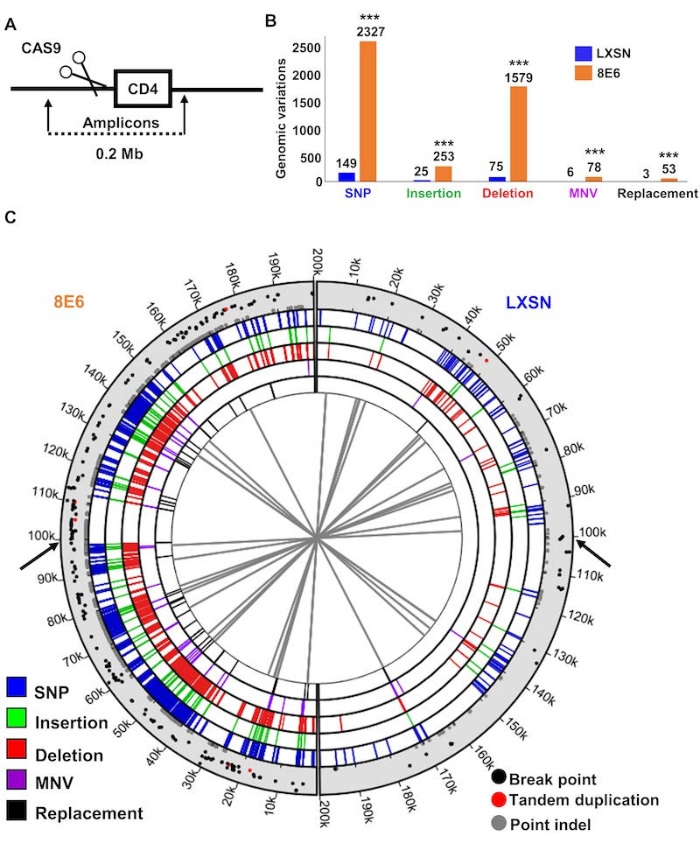
Figure 3: Beta-HPV 8E6 increases genomic instability during DSB repair. (A) Schematic of the placement of CAS9 induced DSB along the sequenced portion of the genome. (B) Genomic variations grouped by types of mutational events in HFK LXSN and HFK 8E6. Each group of genomic variations and total number of variations were compared between HFK LXSN and HFK 8E6. (C) Circos plot of DNA mutations in HFK LXSN (right side) and HFK 8E6 cells (left side). Black arrows indicate CAS9 cutting sites. The innermost circle displays connections between identical genomic rearrangements. The location of genomic rearrangements colored by types of genomic variations are shown in five concentric circles (blue represents SNP, green represents insertion, red represents deletion, purple represents MNV, and black represents replacement). Scatter plot in the outermost circle displays breakpoints (black), tandem duplications (red), and point indels (gray), in which the proximity to the outer edge represents high variant ratio. SNP, single nucleotide polymorphism. MNV, multi-nucleotide variation. Statistical differences between cell lines were measured using a Students' t-test. *** indicates p < 0.001". This is adapted from a previously published reference with permission22. Please click here to view a larger version of this figure.
Table 1: PCR Master mix components. Please click here to download this Table.
Table 2: PCR Program settings. Please click here to download this Table.
Table 3: Trouble shootings. Please click here to download this Table.
Supplemental Table 1: List of primer pool for PCR. Please click here to download this Table.
