Cancer is caused by the abnormal growth of normal cells and is one of the most lethal diseases around the world. These abnormal cells spread to different organs in the body by a process called metastasis. The most common form of cancer is breast cancer, which occurred in 2.26 million people in 2020. Moreover, there were around 1.80 million deaths due to lung cancer in 20201. According to the World Health Organization, around 10 million people died from cancer in 20202. Cancer cells differ from normal cells in that they overexpress certain enzymes, such as protein tyrosine kinases (PTKs). The National Cancer Institute defines kinases as enzymes able to phosphorylate other proteins or sugars3. Knowledge of the regulatory function of kinases can facilitate the design of effective anticancer drugs. For example, PTKs catalyze the phosphorylation of other proteins or sugars, and as a consequence, ATP is converted to ADP by the loss of a phosphate group. A total of 80% of oncogenes and protooncogenes encode PTKs4. Src kinases are a family of non-receptor tyrosine kinases, including Lck, Fyn, Hck, Blk, Yes, and Yrk, that are overexpressed in cancer cells, especially in breast cancer5,6. Src tyrosine kinases are associated with mitogenesis, differentiation, T-cell activation, and cell transformation. Src helps cancer cell invasion and metastasis due to its ability to reduce cancer cell adhesion. There are five different domains in Src kinase, ordered from the N- to C-terminals as: fatty acid domain, Src homology 3 domain (SH3), Src homology 2 domain (SH2), tyrosine kinase domain (SH1), and C-terminal regulatory domain7.
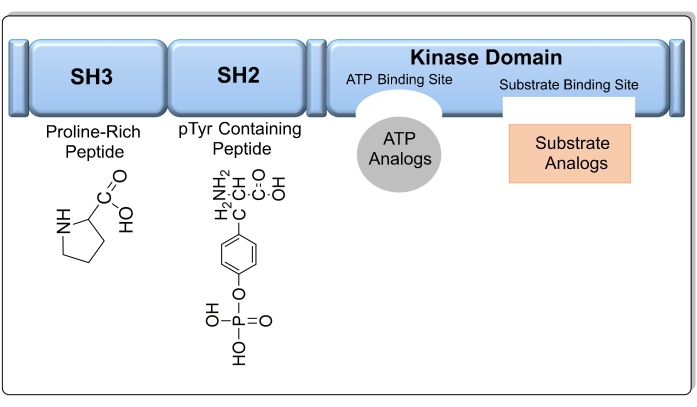
Figure 1: The target domains in the Src kinase enzyme, including a SH3 domain, SH2 domain, kinase domain (SH1), and a short C-terminal regulatory segment. Please click here to view a larger version of this figure.
The kinase domain SH1 is most commonly targeted when designing Src kinase inhibitors, as it contains two conserved sites for ATP and substrate binding (Figure 1). If the amino acid sequence of the kinase domain is known, the substrate can also be used as a target to design a compound that mimics substrate binding to Src kinase8. In addition, other sites such as the SH3 and SH2 domains can be used as targets. Compared to other chemotherapy agents, kinase inhibitors exhibit less toxicity and higher efficacy9. As of September 2021, there are 73 small molecules that act as kinase inhibitors that have been approved by the FDA10. Imatinib is an example of an anticancer drug that selectively inhibits the activity of tyrosine kinase; however, some patients are resistant to the drug due to the appearance of a point mutation in the kinase domain11. AstraZeneca released Saracatinib, which is a drug that inhibits the Src family of tyrosine kinases with an IC50 value (the concentration at which 50% inhibition occurs) of 2.7 nM, but it was discounted in phase 2 trials12. Of the 52 PTK inhibitors approved by the US FDA as of the beginning of 202013, only 28 target receptor PTKs, 11 block the non-receptor PTK, 11 inhibit protein-serine/threonine protein kinases, and two block MEK1/213. The increasing research interest in oncology will continue to fuel the discovery of kinase inhibitors as potential anti-cancer drugs. However, only 50 out of 500 protein kinases have been targeted for treatment thus far; therefore, a greater number of kinases are expected to be studied for drug development in the near future14. In addition, there is a need to discover kinase inhibitors to explore as yet unidentified kinase mutations that lead to cancer.
Thus, this study aimed to develop peptides that could be used as inhibitors for the Src family and target the ATP binding site due to its ability to serve as a conserved site between different kinases. To this end, a series of dipeptides containing methylated tryptophan and/or methylated arginine were synthesized and tested for their synergistic ability to inhibit Src kinase. The indole ring of tryptophan mimics the adenine of ATP and competes with ATP from binding to the ATP-binding site. In addition, the methylated arginine in the ligand competes for the SH3 domain of Src. Researchers showed that a polypeptide containing demethylated arginine inhibits the SH3 domain, possibly due to a specific conserved sequence on the SH3 binding motif (i.e., PXXP), which has a binding affinity to a ligand containing two to three Arg residues in the N-terminal or one to two Arg residues on the C-terminal of the ligands15,16,17. The guanidino group of Arg binds to the conserved Asp-99 residue of the SH3 domain18,19, while the remaining portion of the Arg binds to the conserved Trp-118 of the enzyme, as confirmed from NMR analysis and the crystal structures of several SH3 domains19. Here, a protocol for the synthesis of seven methylated dipeptides and testing their inhibition ability against Src kinase is presented. Further, the ability of these peptides to kill several cancer cell lines in vitro was examined.
