The method described here was successfully applied to visualize intraspecies competition in a cell population consisting of B. subtilis strains that were labeled with the cfp and yfp genes encoding the fluorophores CFP and YFP, respectively. As shown in Figure 3, the method can be used to visualize intraspecies competition in a very illustrative manner. By spotting the samples on small areas, the clonal composition of the cell population was made visible at a glance. Although not appropriate for quantitative analyses, this approach is useful for roughly estimating the effect of different growth parameters (i.e. nitrogen source) on the development of a cell population that initially contained both strains in equal amounts (Figure 3). Moreover, in a small-scale approach the fitness of different B. subtilis strains that were cultivated under the same growth condition can be tested using a single agar plate. For quantitative analyses it is recommended to propagate the samples over the whole surface of an agar plate. This will prevent overlay of the colonies and thus allows the distinct identification and count of colonies that emerged from single cells. By plating appropriate dilutions on agar plates the clonal composition of a cell population over time can be precisely determined simply by counting the yellow and blue fluorescent colonies (see Figure 4). As we have previously reported, GDH activity strongly affects fitness of B. subtilis depending on the availability of exogenous glutamate2. Obviously, in the absence of exogenous glutamate high-level GDH activity is disadvantageous for the bacteria as the enzymes RocG and GudB degrade glutamate that is needed in anabolism (see Figure 1 and Figure 4A). By contrast, if provided to the bacteria, glutamate can serve as an amino group donor in transamination reactions. Moreover, glutamate can be fed into carbon metabolism and used as a source of energy due to the presence of the catabolically active GDHs RocG and GudB (Figure 1 and Figure 4B). As shown in Figures 4C and 4D, similar results were obtained in a dye-switch experiment. Again, bacteria equipped with high-level GDH activity were outcompeted by cells with reduced GDH activity in growth media lacking glutamate. By contrast, bacteria synthesizing only one active GDH were eliminated from the culture when the medium was supplemented with glutamate. As shown in Figures 5A and 5B, the initial composition of the mixed cell population remained almost constant over time. Thus, in the competition experiment the elimination of either of the two strains that were equipped with different amounts of GDH activity was not due to a growth defect caused by the fluorophores (see Figure 4). Taken together, the usage of fluorophores is a powerful tool for monitoring intraspecies competition in a bacterial cell population.
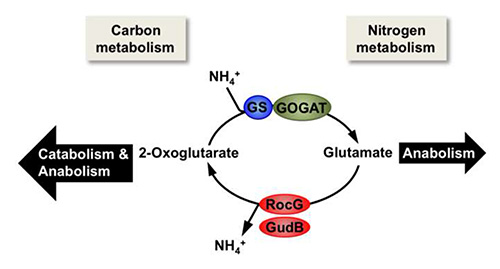
Figure 1. The link between carbon and nitrogen metabolism in B. subtilis. When glutamate is not provided by the medium, the major amino donor that is needed for anabolism is synthesized from ammonium and 2-oxoglutarate by the combined action of the glutamine synthetase (GS) and the glutamate synthase (GOGAT). By contrast, in the presence of exogenous glutamate the catabolically active GDHs RocG and/or GudB can degrade glutamate to ammonium and 2-oxoglutarate, which then serves as a carbon source.
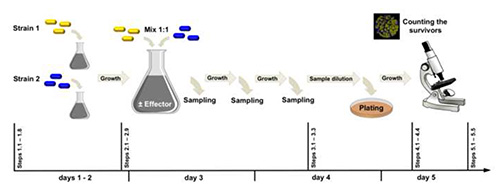
Figure 2. Experimental workflow. Strain 1 (labeled with yfp) and strain 2 (labeled with cfp) differ in one locus from each other. In the example presented here, we have compared the effect of exogenous glutamate (effector) on the genotypic shift of the cell population that initially contained 50% of rocG+ gudB+ (encoding two active GDHs) and 50% of rocG+ gudBCR (encoding one active GDH) cells. Click here to view larger image.
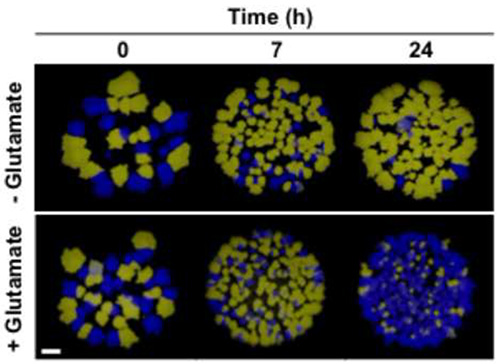
Figure 3. Semiquantitative approach to visualize intraspecies competition in a descriptive way (see section 5). Prior to cocultivation (0 hr), and after 7 hr and 24 hr of growth dilutions (10-4) of cells were spotted on SP agar plates. The surviving cells that have formed colonies after 12 hr of incubation at 37 °C were identified by stereo fluorescence microscopy. Exposure time, 0.6 sec; scale bar, 1 mm. This figure was modified from Gunka et al. 20132.
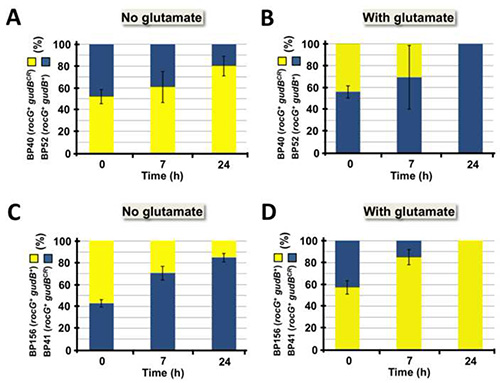
Figure 4. Quantification of intraspecies competition. After sample dilution and propagating the cells (see steps 3.1-3.3) on SP medium the plates were incubated overnight at 37 °C. Yellow and blue colonies were quantified as described in Protocols 4 and 6. The black error bars represent standard deviations for at least four independently repeated experiments. Each agar plate contained at least 100 countable colonies. (A) In the absence of exogenous glutamate the B. subtilis strain BP40 (yellow) equipped with only one functional GDH outcompetes strain BP54 (blue), which synthesizes both glutamate-degrading enzymes, RocG and GudB. (B) By contrast, synthesis of two functional GDHs is advantageous for the bacteria when exogenous glutamate is available because in addition to glucose, glutamate is used as a carbon source. As shown in (C) and (D), comparable results were obtained in a dye switch experiment. This figure was modified from Gunka et al. 20132. Click here to view larger image.
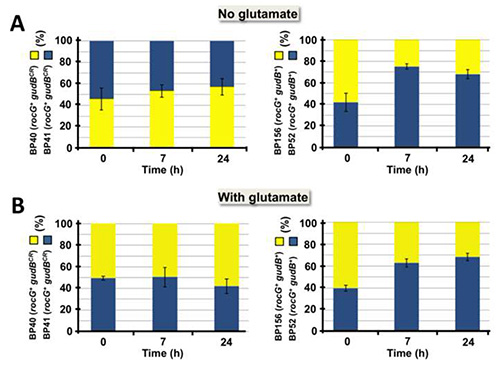
Figure 5. Control experiment to evaluate the effect of the fluorophore-encoding cfp and yfp genes on fitness of the bacteria. Mixed populations of the isogenic strains BP40 (rocG+ gudBCR amyE::yfp) and BP41 (rocG+ gudBCR amyE::cfp) or BP52 (rocG+ gudB+ amyE::cfp) and BP156 (rocG+ gudB+ amyE::yfp) were grown in the absence (A) and in the presence (B) of exogenous glutamate. The surviving cells were counted as described in Protocols 1-4 and 6, respectively. The bars represent standard deviations for at least four independently repeated experiments. This figure was modified from Gunka et al. 20132. Click here to view larger image.
