Figure 1 represents a typical UV/Vis spectrum of RNA isolated from serum. From this profile we noted protein contamination at 280 nm with phenol and organic contaminants both at 270 nm and 230 nm, respectively. Residue guanidinium thiocyanate was also noted at 260 nm. To reduce contaminants, a series of optimization steps were made to the standard Tri-Reagent RT-LS procedure. We added 5 mg/ml of glycogen to increase both the total RNA yield and reduce contamination (black line, Figure 1A).
Further we observed that after the removal of isopropanol there was roughly 100-300 µl of wash buffer surrounding the RNA pellet. An additional centrifuge spin was added to the protocol and the residue wash solution was carefully removed. This resulted in a profile shift from 270 nm to 280 nm (red trace, Figure 1B). We deduced that this "flash" spin had reduced the phenol contamination at 270 nm and concluded that the 280 nm peak most likely represented protein contamination.
To further increase RNA yield and reduce protein carry through we added a Phase Lock Gel step. This step allowed for the complete transfer of the aqueous phase without organic contaminants (Figure 1B). As serum contains a large abundance of protein, we evaluated if diluting the original serum volume would make any difference (Figure 1C). In a total extract volume of 500 µl, we mixed 400 and 250 µl of serum with dH2O. The larger volume yield nearly doubled the amount of total RNA when compare to using 250 µl of serum. There was also a reduction in contaminants when decreasing the starting volume (Note: As the only variable in this optimization step was the volume of serum, the 230 nm peak is directly associated with serum volumes and not an artifact from the Tri-Reagent isolation).
The small RNA content of these preparations was then assessed using the Bioanalyzer in combination with the Small RNA Kit. From the Bioanalyzer trace, there is a distinct peak at approximately 20 nts which represents the microRNA population (Figure 2A). Using this protocol, this microRNA component represented 93% of the total small RNA population. This is a high level of purity and the total RNA can now be used for varying molecular assays such as arrays and qPCR reactions. A summary of the workflow is presented in Figure 2B.
We then measured microRNA expression in four different biological samples using either an oligonucleotide array or qPCR. Figure 3A represents a microRNA heat map of these four samples. As in our previous study, hierarchical clustering was used to analyze the expression data and group samples based on their microRNA expression profile8. Quantitative PCR (qPCR) was also used to assess microRNA levels in these preparations. Using the same four samples, we performed singleplex TaqMan qPCR reactions to detect miR-21-5p, miR-486-5p, miR-15b-5p, miR-16-5p and let-7a. As shown in the amplification plots (Figures 3B and 3C) all miRNAs were successfully detected.
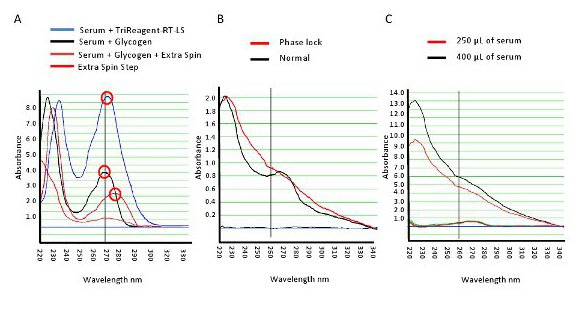
Figure 1. Spectrophotometric profile of RNA from human serum. A. A typical UV profile of RNA isolated from human serum (blue line). Various optimization steps were tested to reduce contaminates and increase total RNA yield. B. Compares the use of a Phase Lock Gel to a normal isolation. C. Profile of two measurements with different starting volumes of serum. Increasing the volume had marked impact on RNA yields. Please click here to view a larger version of this figure.
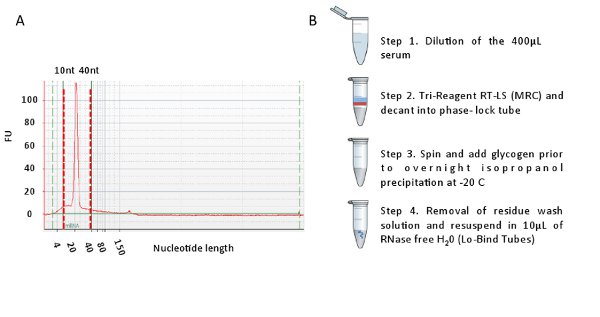
Figure 2. A. Representative Bioanalyzer trace showing a single spike at approximately 21 nucleotides (x-axis). This spike represents the miRNA fraction in the small RNA population from serum. B. Summary of the workflow to isolate total RNA from serum. Please click here to view a larger version of this figure.
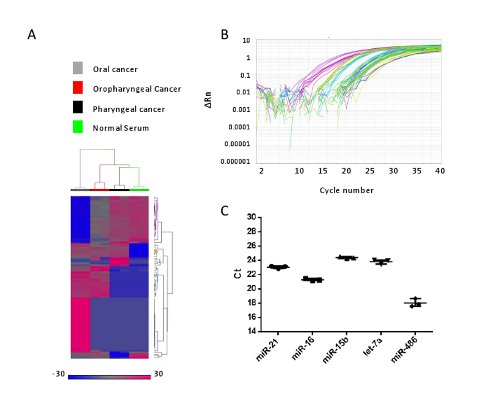
Figure 3. The microRNAs isolated using this method were utilized for array profiling and qPCR analysis. A. Three head and neck cancers and one normal sera were isolated for total RNA (including microRNAs) and subjected to array profiling using the 8 X 60K miRNA chip. This was repeated in technical replicates. After raw data Quality Control (QC) processing, expression of these microRNAs was analyzed using Hierarchical Clustering (HCL) and presented as a heat map. Red indicates up regulation and blue indicates down regulation of the microRNAs. B. Representative qPCR amplification curves for the detection of miR-21, miR-486, miR-15, miR-16 and let-7a in four human sera. C. All five miRNAs were detected and raw Ct values were then plotted for the normal serum. This pattern of detection was similar for the other three samples (Data not shown). Please click here to view a larger version of this figure.
