Using the protocol outlined within, we present representative data demonstrating flow cytometric detection of ROS production resulting from stimulation of WT C57BL/6J BMDMs through the FcγR. As expected, we observe minimal changes in FL1 or FL2 fluorescence above background levels in unstimulated cells (Figure 3A, compare “stained, unstimulated” vs “unstained, unstimulated” dot plots). We observe a marked increase in FL1 and FL2 fluorescence when cells are stimulated with FcγR cross-linking agent (Figure 3A, compare “stained and stimulated via Fc cross-linking” samples vs “stained, unstimulated” dot plots). Lastly, when cells were treated with ROS inhibitor prior to FcγR cross-linking, this increased fluorescence is brought back to basal levels (Figure 3A, compare “stained and treated with ROS inhibitor and stimulated via Fc cross-linking” vs “stained and stimulated via Fc cross-linking” dot plots). This is also evident when data is presented as a histogram for each channel (Figure 3B) or when data is presented as a percentage of cells positive for either the green or orange ROS probes (Figure 3C). A similar trend is also apparent when data is presented as MFI, although the reduction in orange fluorescence with pre-treatment with ROS inhibitor is not captured as well when presented as MFI versus as a percentage (Figure 3C). We also present the results of 3 independent experiments performed on different days (Figure 3, Experiment 1, 2, and 3). The average values and corresponding standard error of the mean are indicated in the graphs (Figure 3C).
We also present unsuccessful experimentation, where sub-optimal ROS production as a result of FcγR stimulation was observed (Figure 4). A minimal increase in FL1 and FL2 fluorescence was detected when comparing “stained and stimulated via Fc cross-linking” samples vs “stained, unstimulated” samples (Figure 4A,B,C). This is presented alongside a successful experiment to highlight the large differences between the expected percentages or MFI increases and the observed values in the unsuccessful experiment.
The current protocol utilizes a 24 h priming step. When comparing a 24 h versus a 48 h priming time, we observed no marked difference in the percentage of cells positive for the green, oxidative stress reagent (Figure 5A, top histograms and Figure 5B green probe, % positive). However, increasing the priming time to 48 h did increase the percentage of cells positive for the orange fluorescence (Figure 5A, lower histograms and Figure 5B orange probe, % positive). This was similarly reflected when data was presented as MFI. This suggests that for optimal detection of all ROS species, a 48 h priming time may be more ideal.
Given that, due to the cost or the time needed for experimentation, use of a kit to perform this assay may not be an option. For this reason, we also tested similar components to those provided in the kit and purchased these from standard vendors (Thermofisher, EMD Millipore, Cayman). We find that using individually procured components and the same experimental protocol for cell loading and FcγR stimulation, we can recapitulate many of the same findings we observed using the kit (Figure 6A,B). However, although increases in fluorescence were apparent with stimulation, a higher level of ROS production was observed using the kit. This may indicate that use of individually procured components may be feasible but would need to be further optimized for this specific assay.
Lastly, we demonstrate that it is also possible to combine cell-surface staining with these ROS probes. We use a known macrophage marker, F4/80, conjugated to Alexa 647 and perform cell surface staining prior to treatment with ROS inhibitor, ROS inducer, or specific stimuli to induce ROS production. We demonstrate in Figure 7B that macrophages respond as expected when treated with FcγR crosslinking agent and FcγR crosslinking agent + ROS inhibitor (Figure 7B, orange vs green dot plots). Furthermore, we can observe increased orange or green fluorescence specifically generated by the F4/80 labeled cells upon treatment with FcγR crosslinking agent and reduced with pre-treatment with ROS inhibitor (Figure 7B, F4/80 vs green and F4/80 vs orange dot plots).
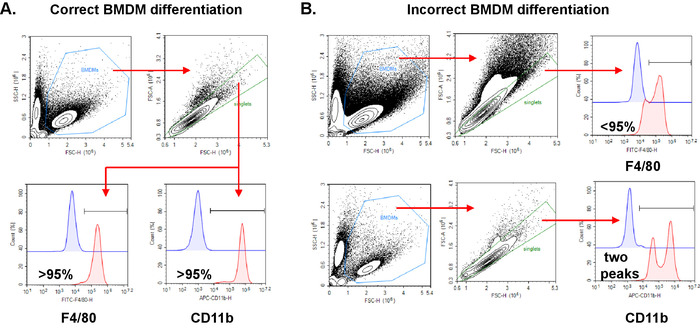
Figure 1: Flow cytometric assessment of appropriate generation of BMDMs. Wild type BMDMs were generated and were left either unstained or stained with FITC anti-mouse F4/80 or Alexa 647 anti-mouse CD11b. FSC (x-axis) vs SSC (y-axis) plots were generated and macrophages (BMDMs) were gated to exclude dead cells and debris. Using a plot of FSC-H(x-axis) vs FSC-A (y-axis), a singlet gate was generated. Gating on singlets, histograms were generated to show cells stained with either FITC F4/80 or APC CD11b in comparison to the isotype stained control. A) Correct BMDM differentiation with more than 95% of cells staining positive for CD11b or F4/80. B) Incorrect BMDM differentiation where less than 95% of cells are staining positive for F4/80 and 2 peaks are present for CD11b. Please click here to view a larger version of this figure.
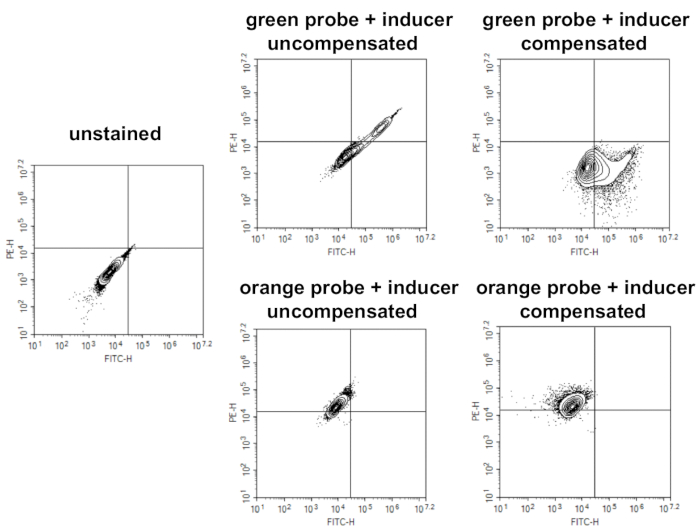
Figure 2. Performing compensation when using green and orange ROS probes. Compensation will require unstained untreated cells, cells stained with green ROS probe and treated with ROS inducer, and cells stained with orange ROS probe and treated with ROS inducer. Dot plots for unstained untreated cells are used to determine quadrant gates. Data for cells singly stained with either green ROS probe or orange ROS probe and treated with ROS inducer are shown prior to, and after, compensation was applied. The compensation matrix is then applied to all subsequent experimental samples. Please click here to view a larger version of this figure.
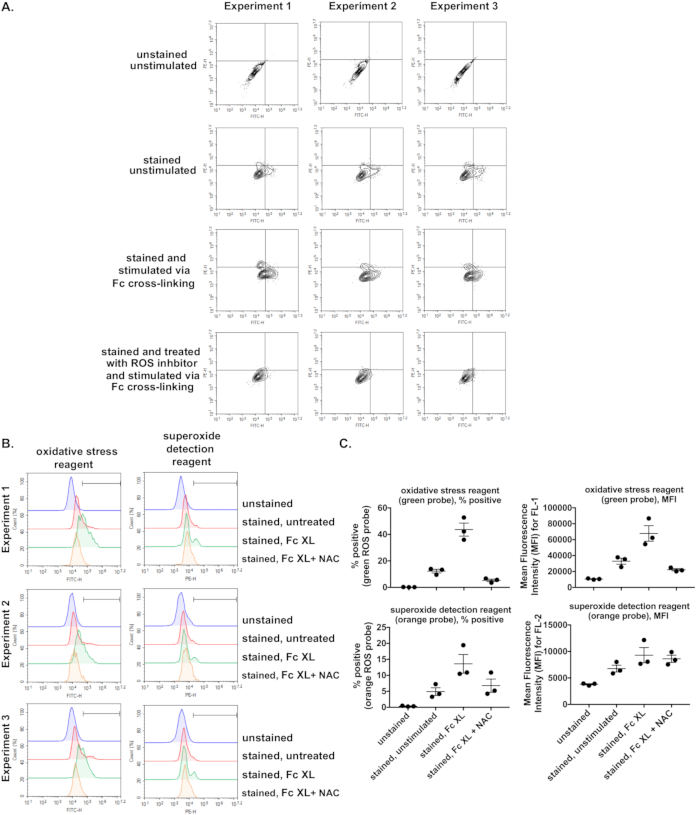
Figure 3. Measurement of ROS in response to specific FcγR stimulation using green and orange ROS probes and assessment of assay reproducibility. Wild-type bone marrow-derived macrophages (BMDMs) were generated, primed, and were left either unstained or stained with a cocktail of green and orange ROS probes. Stained BMDMs were either left untreated, stimulated through their FcγRs using murine anti-BSA IgG1 + BSA for 30 min, or treated with ROS inhibitor prior to stimulation via FcγR cross-linking. A) Dot plots, showing an increase in the percentages of cells in the upper left, upper right, and lower right quadrants upon specific FcγR stimulation, which is reduced in the presence of ROS inhibitor. B) Histograms of each fluorescence channel, showing the marker gate to determine cells positive for each probe. C) Presentation of the data as a percentage of cells positive for each probe or as an increase in MFI. Three independent, representative experiments are presented. The mean for the 3 experiments and standard errors of the mean are shown as lines within the graphs. Fc XL, Fc cross-linking; NAC, N-acetyl-L-cysteine. Please click here to view a larger version of this figure.
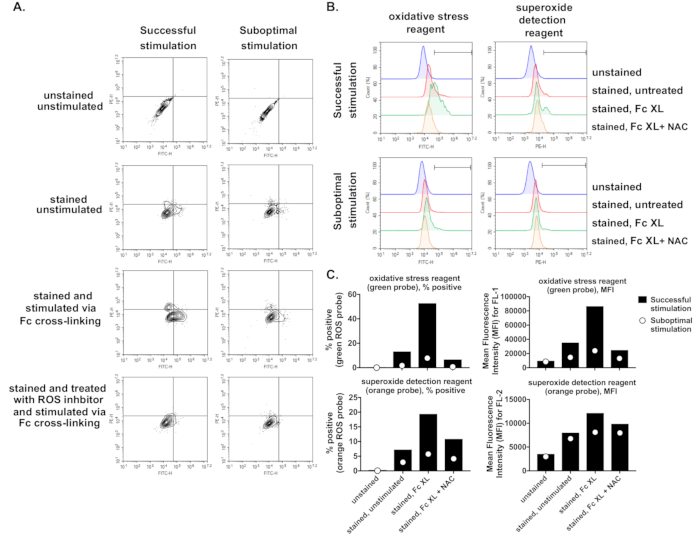
Figure 4. Examples of successful and suboptimal FcγR stimulation. Wild-type bone marrow-derived macrophages (BMDMs) were generated, primed, and were left either unstained or stained with a cocktail of green and orange ROS probes. Stained BMDMs were either left untreated, stimulated through their FcγRs using murine anti-BSA IgG1 + BSA for 30 min, or treated with ROS inhibitor prior to stimulation via FcγR cross-linking. Representative results for successful and suboptimal stimulation are shown as A) dot plots, B) histograms, or C) the percentage of cells positive for each probe or as an increase in MFI. Successful stimulation shows increased fluorescence in the upper left, upper right and lower right quadrants of the FL1 vs FL2 plot and increased MFI and percentage of positive cells stained with each probe upon FcγR stimulation. Suboptimal stimulation shows minimal increase in MFI or percentage of positive cells. Fc XL, Fc cross-linking; NAC, N-acetyl-L-cysteine. Please click here to view a larger version of this figure.
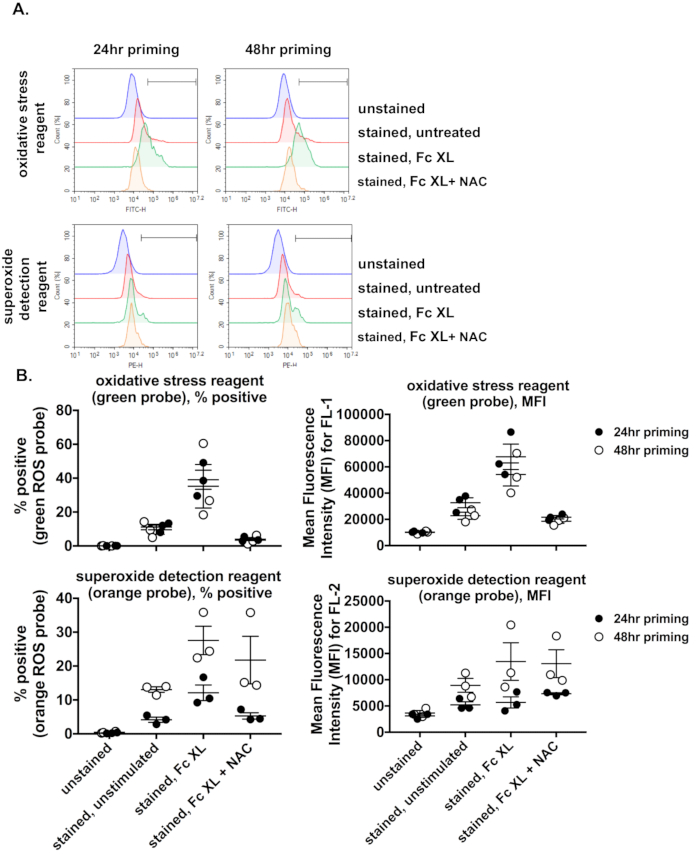
Figure 5. Effect of priming time on ROS generation upon FcγR stimulation. BMDMs were generated, primed for either 24 or 48 h, and were left either unstained or stained with a cocktail of green and orange ROS probes. Stained BMDMs were either left untreated, stimulated through their FcγRs using murine anti-BSA IgG1 + BSA for 30 min, or treated with ROS inhibitor prior to stimulation via FcγR cross-linking. A) Histograms for the fluorescence induced by each probe upon stimulation of macrophages primed for either 24 or 48 h. B) Percentage of cells positive for each probe upon stimulation of macrophages primed for either 24 or 48 h. Priming macrophages for 48 h resulted in an increase in percent of cells positive for orange fluorescence (or an increase in the MFI for the FL2 channel) compared to priming the macrophages for 24 h. The mean for the 3 experiments and standard errors of the mean are shown as lines within the graphs. Fc XL, Fc cross-linking; NAC, N-acetyl-L-cysteine. Please click here to view a larger version of this figure.
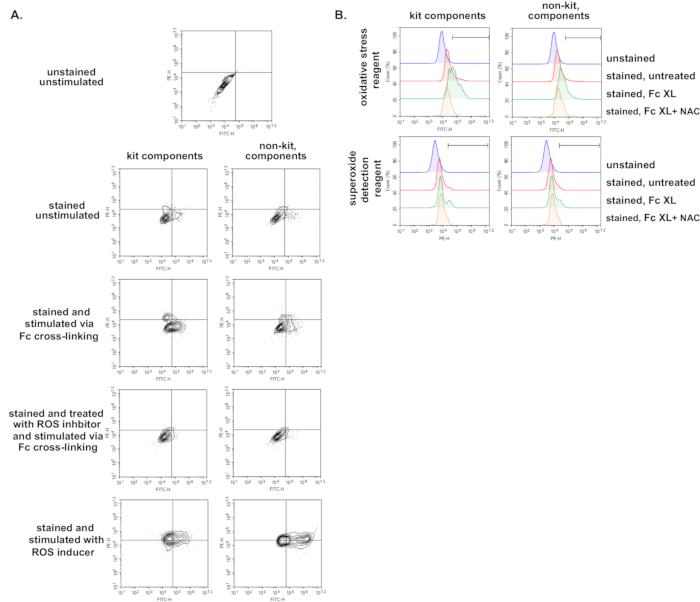
Figure 6. Flow cytometric ROS measurement upon FcγR cross-linking using reagents from different vendors. BMDMs were generated, primed for 24 h, and were left either unstained or stained with a cocktail of oxidative stress and superoxide detection probes. Stained BMDMs were either left untreated, stimulated with ROS inducer, stimulated through their FcγRs using murine anti-BSA IgG1 + BSA for 30 min, or treated with ROS inhibitor prior to stimulation via FcγR cross-linking. Probes, ROS inducer and ROS inhibitor were used either from the kit or were purchased separately from different vendors and used at a similar concentration. A) Dot plots showing a side-by-side comparison of results obtained using kit and non-kit components. B) Histograms showing a side-by-side comparison of results obtained using kit and non-kit components. Fc XL, Fc cross-linking; NAC, N-acetyl-L-cysteine. Please click here to view a larger version of this figure.
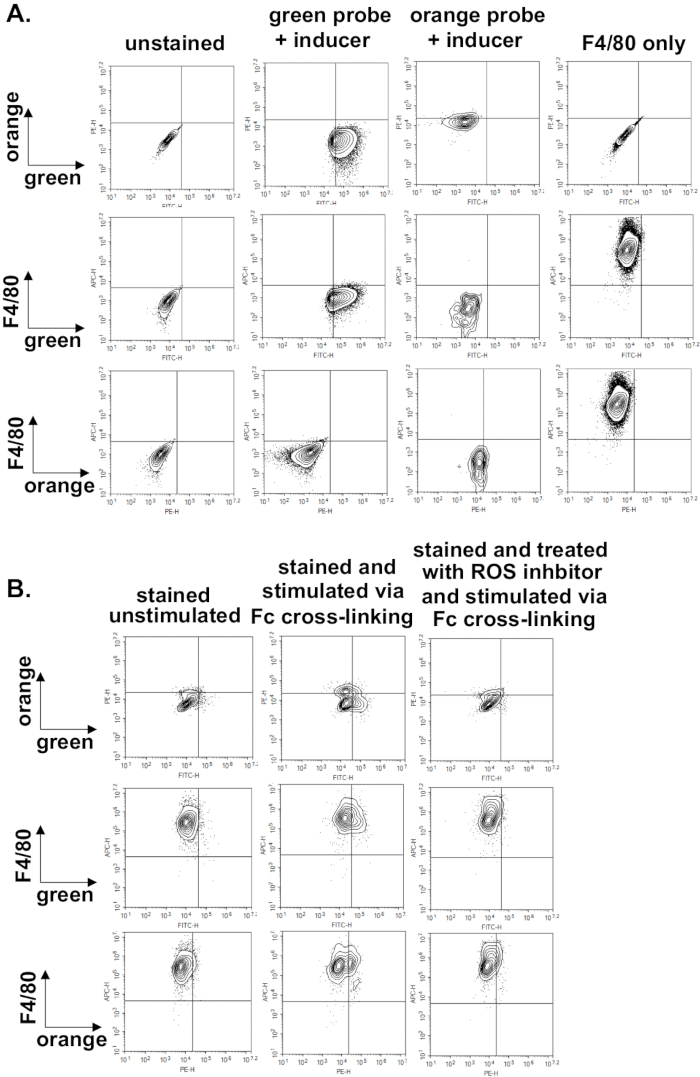
Figure 7. Combining cell-surface staining with flow cytometric measurement of ROS production upon FcγR cross-linking. A) Dot plots for the various fluorescent channels using unstained or singly stained compensation controls. Wild-type BMDMs were generated, primed for 24 h, and were left either unstained, stained with Alexa 647 anti-mouse F4/80 only, stained with green ROS probe only and treated with ROS inducer, or stained with orange ROS probe only and treated with ROS inducer. Dot plots for unstained untreated cells are used to determine quadrant gates. Plots demonstrate expected results if channels are correctly compensated. The compensation matrix was then applied to all subsequent experimental samples. B) Wild-type BMDMs were generated, primed for 24 h and stained with Alexa 647 anti-mouse F4/80. Afterward, BMDMs were either left unstimulated, stimulated through their FcγRs using murine anti-BSA IgG1 + BSA for 30 min, or treated with ROS inhibitor prior to stimulation via FcγR cross-linking. Dot plots demonstrate that green or orange fluorescence specifically produced by F4/80 cells can be detected. Please click here to view a larger version of this figure.
