新一代测序为可操作的突变在固体和液体瘤检测
Summary
This manuscript describes clinical protocols for two next-generation sequencing panels. One panel interrogates hematologic malignancies while the other panel targets genes commonly mutated in solid tumors. Molecular classification of driver mutations in human malignancies offers valuable prognostic and predictive information.
Abstract
As our understanding of the driver mutations necessary for initiation and progression of cancers improves, we gain critical information on how specific molecular profiles of a tumor may predict responsiveness to therapeutic agents or provide knowledge about prognosis. At our institution a tumor genotyping program was established as part of routine clinical care, screening both hematologic and solid tumors for a wide spectrum of mutations using two next-generation sequencing (NGS) panels: a custom, 33 gene hematological malignancies panel for use with peripheral blood and bone marrow, and a commercially produced solid tumor panel for use with formalin-fixed paraffin-embedded tissue that targets 47 genes commonly mutated in cancer. Our workflow includes a pathologist review of the biopsy to ensure there is adequate amount of tumor for the assay followed by customized DNA extraction is performed on the specimen. Quality control of the specimen includes steps for quantity, quality and integrity and only after the extracted DNA passes these metrics an amplicon library is generated and sequenced. The resulting data is analyzed through an in-house bioinformatics pipeline and the variants are reviewed and interpreted for pathogenicity. Here we provide a snapshot of the utility of each panel using two clinical cases to provide insight into how a well-designed NGS workflow can contribute to optimizing clinical outcomes.
Introduction
临床肿瘤学标本下一代测序(NGS),为查明靶向基因变化和预测/预后分子标记的重要性越来越多的科学文献点已经成为过去几年更广泛的应用。多基因分析小组和疾病进展和复发两种上皮1,2和血液系统恶性肿瘤3全外显子组测序研究都凝固了肿瘤异质性的概念,并克隆演变。另外,与竞争技术如聚合酶链反应(PCR)或Sanger测序,NGS可以检测单个测定4中的所有临床相关的癌基因最基因组改变。
该中心最初有两个临床NGS板,自定义面板血液学(血红素-NGS面板)和一个现成的,货架癌症小组FFPE样品(固体NGS面板)推出个性化诊断(见<strong>图1)。这些面板覆盖了选择的基因的临床相关的或高利息的区域;不是所有的基因或外显子完全覆盖。扩增子被探针杂交其次是延伸和连接产生的。目标区域使用PCR具有普遍的双索引引物进一步放大,允许多达96个样品进行汇集测序。
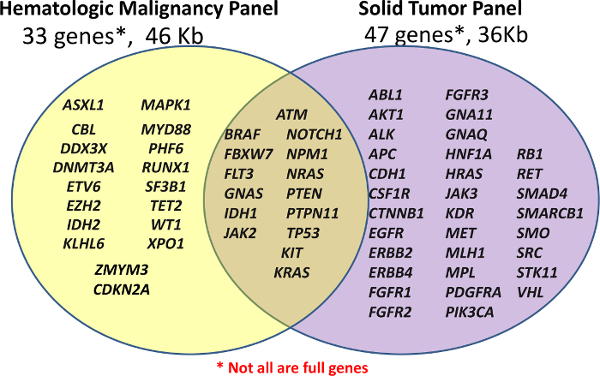
图1:在面板覆盖基因的列表中使用进行文库制备为自定义面板血液学33基因或关闭的,现成的扩增子癌症面板的47个基因(固NGS)(血红素-NGS面板)。不是所有的基因或外显子的覆盖全面,因为有些可能扩增子仅涵盖某些热点。 请点击此处查看该图的放大版本。</ A>
为血红素-NGS面板内容是从多个来源,但围绕在先前描述为证明临床效用5高水平急性髓细胞白血病(AML)突变16个基因中心。固NGS面板与商业报道的体细胞突变的目录癌症(COSMIC)数据库6基于在一般癌症的基因突变目标区域产生的。
几个关键步骤表征整个工作流程,为临床NGS。临床医生订单试验后,病理学家确定以下用于肿瘤百分比和样品体积分析检体的充分性。在我们的机构,我们要求,由于该技术的背景测序错误率(“噪声”)和目标方法的效率为至少10%的肿瘤。如果组织是足够的检测,基因组DNA提取。然后将该DNA进行多个质量控制(QC)的步骤。如果DNA通过QC,产生和测序的扩增子文库。所得数据通过一个内部的生物信息学流水线进行分析。继生物信息学分析,变种人工审查,并结合之前解释的致病性到临床报告。下面我们描述了两个案例,通过这个严格的流程去,并最终导致临床管理的变化。
案例1 -急性髓系白血病
从病人A A骨髓活检诊断是反洗钱,没有成熟。细胞遗传学研究是对骨髓标本发出并表现出正常的雌性核型。出席者达95%循环爆炸,使外周血标本为血红素NGS面板上的个性化诊断测试发送。
急性髓性白血病(AML)是白血细胞的骨髓谱系的血液系统恶性肿瘤。检测在AML基因突变已成为预后和治疗越来越重要,复发性基因突变公认的发病机制和预后7重要。在NPM1和CEBPA突变与一个有利的预后风险相关,而在FLT3内部串联重复(场ITD)已与一个不太有利的结果8有关。越来越多的证据体支持的AML 9这些和其他突变的致病作用。
案例2 -肺腺癌
从患者B中的左锁骨上肿块活检证实肺腺癌。从福尔马林固定的石蜡包埋(FFPE)淋巴结肿块活检材料用于基因组试验(固NGS面板)被发送作为辊/卷发具有大于50%的肿瘤,以确定一个突变是否存在有针对性的治疗干预。
龙CANC器是在美国癌症相关死亡的主要原因,并分为两种主要类型,非小细胞肺癌(NSCLC)和小细胞肺癌(SCLC)。非小细胞肺癌,可以进一步定义为腺癌或鳞状细胞癌,基于病变的组织学。肺腺癌是肺癌最常见的亚型,在吸烟者和非吸烟者看到的,是不吸烟者10肺癌最常见的形式。肺腺癌的分子研究已经在多个癌基因11中确定突变。在吸烟者中确定的最常见驾驶员的突变是在KRAS基因和BRAF突变。在非吸烟者中最常见的突变是EGFR突变,以及涉及的基因ALK,RET和ROS1重排。肺肿瘤已与一个在框架的外显子20插入在基因ERBB2(HER2 / neu的)中描述。在何最常见的异常R 2 / neu的是该位点在乳腺癌的扩增为其中靶向治疗是可用的(曲妥单抗:针对HER2 / neu基因的人源化单克隆抗体)。即在2观察HER2 / neu基因外显子20插入-肺4%adenocarcimomas 12已经(分别为那替尼和西罗莫司脂化)13显示,联合治疗HER2 / neu和mTOR抑制剂部分缓解。
Protocol
Representative Results
Discussion
如本手稿描述的两个NGS测试临床上所提供的最重要的实际考虑是质量控制。具体来说,仔细考虑必须支付的质量和提取的DNA量。这是为FFPE样品这往往是用可变的DNA产量高度退化尤其重要。异丙醇沉淀法,是为了最大限度地从FFPE样品DNA产率,为发现基于列的方法有时会导致有限的洗脱体积的DNA剪切开发。因此,大多数的当检体产生浓度太低或为测定太降解的时间,这是最有可能是由于该组织的大小,类型,或固定和不提取过程。对血液/骨髓样本,如果有一个提取失败,它通常是由于样品被hemodilute( 即不具有足够数目的白细胞或肿瘤细胞在该拉伸的)或化学消融。
。NT“>验证期间,进行DNA的质量和数量的可接受性截断应建立100的推荐输入 – 250纳克在测定通常使用;但是,如果DNA质量是好的,那么较低的输入量可以是成功的。此外,如果该DNA质量差( 即 ,扩增的DNA的量小于100 – 250毫微克)然后较高的输入量可以提高测序结果的质量(因为扩增的DNA的量会达到推荐的输入) 。指标DNA的质量和数量应推进DNA插入文库制备之前被应用到每个样本,这些样本在一个“灰色地带”( 见图2)应在实验室负责人或指定人员的自由裁量权运行。目前最好的办法预测如果测序过程中的DNA不会表现良好是执行基于定量PCR的测定法,允许输入DNA的定量和质量评估,这种方法解决了上述bioavailab在样本大小不同的片段,通过不同尺寸的片段( 如 100个基点,150基点,200基点和300基点)和比较收益率的放大ility。目前,文库制备涉及大量的手动步骤,其中在几个接合部之一的失误可导致库失败或以质量较差。微流体凝胶分析是唯一的QC步序之前检查库准备的问题。因此,有几个关键步骤,其中额外留心可以增加成功反应的概率。当务之急是保证正确的样品和寡核苷酸池用于每个样品。确保妥善记录每个样本包含96的一个独特的双索引的PCR引物对的组合,减少了样品混合起来的机会。此外,重要的是要保证过滤板(FPU)正确地排出;如果它不恰当漏这可能会导致EXTE图书馆准备nsion结扎一步次优执行,导致质量差的测序数据。库的QC后,重要的是,确保LNB1珠充分再悬浮并且LNB1 / LNA1溶液是作为该混合物的浓度使用,以确定文库的摩尔浓度将其添加到样品之前充分混合。最后,如果珠洗脱步骤导致库的次优量洗脱离珠粒会降低聚类密度,并可能导致库无法获得足够的平均覆盖率。相反地,过量图书馆,会导致质量较差的读取。因此,要在基于珠子的归一化步骤,以确保定序文库的最佳池和聚类一致是重要的。
除了文库制备,它是验证一个生物信息学管道,将产生从原始的,解复用FASTQ文件准确突变呼叫关键。选择一个因为有许多开源和市售的矫正器,变种呼叫者和NGS软件包,一个人必须要通过筛选定制的解决方案可能是费时。自定义算法将需要设计来提取基本性能统计数据,找出最逃避开源工具独特的经常性突变,并确定在每个位点的拷贝数状态。期间一个生物信息学流水线的验证过程中,重要的是要确定该达到或超过后质量过滤覆盖的两个最小深度( 例如 ,一个最小的250读取)和最小等位基因频率变体的报告的截止值( 例如 ,4个%)。因为这是一个复用基于扩增子的测定法,重要的是要确定最小平均覆盖深度( 例如 ,1000倍),该库需要达到能够得到最低执行扩增到最小深度的读出。此外,该测定的多路复用性质确实Ç澳洲英语脱靶效应和这些“神器”将需要被发现,并在发射前全面审核。所描述的测定法的另一个重要的限制是需要的样品以包含在以达到验证最小等位基因频率大于10%的肿瘤。
低频,1%,FLT3插入的检测是证据表明人工审核仍然在这个过程中所希望的。即使有5%的等位基因频率的截止,一些重要的突变可能错过因而手动审查将是至关重要的,以确定这些变体。对于FLT3-ITD中 ,对所有AML患者进行外显子14目视检查,以确保低级别或大的插入/重复不会被忽视。此外,HER2外显子20插入,其通常是旁边的引物序列,需要人工干预。尽管有强大的生物信息学管道,一些变种可以去忽略它是具有硬砍的只是性质关闭上述大部分的统计数据。将需要更好的生物信息,以帮助缓解这个问题,因为将更好地文库制备和/或测序的方法,因为它是更有利的是含有较少的文物和误报甚至更低截止拥有高质量的数据。
检测和等位基因频率的解释可能是困难的,由于在肿瘤百分比确定和基因组的一些区域的扩增偏差的难度。此外,超过50%的等位基因频率可以被检测,如在壳体2观察到这被解释为杂合事件的损失,无论是由于正常的等位基因的丧失,导致突变体的明显增加读取,一个突变等位基因( 例如 ,2突变体和一个正常复制)或其它机制的增益。这些机制可以通过利用阵列比较基因组杂交(aCGH 19)和/或一个SNP基因分型阵列阐明。20。
目前的目标富集方法依赖于任何低效杂交捕获或多重PCR技术的全日程序,导致需要对单个样品的测序更多的覆盖范围和更多的脱靶序列读取。预期在不久的将来对NGS分子肿瘤额外应用将包括更容易文库的制备方法,可以是完全自动化的,并且能够以非常低的量的DNA模板来处理样品( 即 ,小于1毫微克)以及样品高度降解脱氧核糖核酸。为了应对这些挑战,大多数方法想必会基于PCR的,无论是作为一个多PCR的方法,或大规模并行单一PCR方法。此外,个别的扩增子的分子条形码已经显示出显着降低背景测序噪声,并将使样品的测试与肿瘤细胞的比例较低,以实现较低的等位基因频率并朝向捕获circulat移动ING肿瘤细胞。
在癌组织疾病相关的基因突变的检测一直是护理标准几十年。从历史上看,基因经常一次测试顺序,一个基因/外显子,具有通向测试序列的端部的突变的鉴定。 NGS的出现已经允许偏差较小的方法来测序以并联许多癌症导致了与肿瘤相关的多个突变的标识相关联的多个基因。 NGS的用于癌症检测的体细胞突变的临床应用日益明显。事实上,肿瘤样本的基于NGS-分析代表了一种新的模式,挑战传统的,单一的基因检测,但其临床效用是很清楚的。今天临床实验室有令人兴奋的机会,以结婚认真方法验证和测试的解释与这个强大技术的应用。
Divulgations
The authors have nothing to disclose.
Acknowledgements
作者要感谢丹尼尔野生的援助,用于读取生产的手稿和援助。
Materials
| Genomic DNA ScreenTape | Agilent Technology | 5067-5365 | |
| Genomic DNA Reagents | Agilent Technology | 5067-5366 | |
| High Sensitivity D1000 ScreenTape | Agilent Technology | 5067-5584 | |
| High Sensitivity D1000 Reagents | Agilent Technology | 5067-5585 | |
| TapeStation 2200 | Agilent Technology | G2965A | |
| TapeStation Analysis Software | Agilent Technology | A.01.04 or higher | |
| 96-well Tube Storage Racks | Any Vendor | ||
| 15/50 ml Tube Rack | Any Vendor | ||
| 96-well Plate Rack | Any Vendor | ||
| Pipette, single-channel, 0.5–2.5 μL | Any Vendor | ||
| Pipette, single-channel, 1–10 μL | Any Vendor | ||
| Pipette, single-channel, 2–20 μL | Any Vendor | ||
| Pipette, single-channel, 10–100 μL | Any Vendor | ||
| Pipette, single-channel, 20–200 μL | Any Vendor | ||
| Pipette, single-channel, 100–1000 μL | Any Vendor | ||
| Serological Pipettor | Any Vendor | ||
| Vortexer | Any Vendor | ||
| Ice bucket | Any Vendor | ||
| Microcentrifuge (for tubes and strip tubes) | Any Vendor | ||
| Freezer, -20 °C | Any Vendor | ||
| 4 °C Refrigerator | Any Vendor | ||
| Water or Bead Bath | Any Vendor | ||
| Incubator (37 oC) | Any Vendor | ||
| Serological Pipettes, 1 mL | Any Vendor | ||
| Serological Pipettes, 5 mL | Any Vendor | ||
| Serological Pipettes, 10 mL | Any Vendor | ||
| Serological Pipettes, 25 mL | Any Vendor | ||
| Gloves | Any Vendor | ||
| Razor Blades/Scaples | Any Vendor | ||
| KimWipes | Any Vendor | ||
| 15 mL Conical Tube | Any Vendor | ||
| 50 mL Conical Tube | Any Vendor | ||
| Paper Towels | Any Vendor | ||
| 200 proof Ethanol | Any Vendor | Store in Flammable Cabinet | |
| 2-Propanol (Isopropanol) | Any Vendor | Store in Flammable Cabinet | |
| 25ml Reservoirs | Any Vendor | ||
| 10N NaOH | Any Vendor | ||
| Pipette, 8-channel, 1–10 μL | Any Vendor | ||
| Pipette, 8-channel, 10–100 μL | Any Vendor | ||
| Pipette, 8-channel, 20–300 μL | Any Vendor | ||
| Ice Bucket | Any Vendor | ||
| Water Squirt Bottle | Any Vendor | ||
| Alcohol Squirt Bottle | Any Vendor | ||
| Lens Cleaning Paper | Any Vendor | ||
| Plates, 96-well PCR, Semi-Skirted | Any Vendor | ||
| Tube strips, 8-well, 0.2 mL | Any Vendor | ||
| Agencourt AMPure XP Beads | Beckman Coulter | A63881 | |
| BioShake IQ or 3000-T elm | Bulldog Bio/Q.Instruments | 1808-0506/ 1808-0517 | |
| DropPlate96 S – LabChipDS | Caliper | 128876 | |
| DropPlate96 D – LabChipDS | Caliper | 132848 | |
| DropSense96 | Caliper (Trinean) | ||
| DropQuant Software | Caliper (Trinean) | ||
| Plate Sealing Film | Denville | B1212-5S | |
| Aluminum Seal Foil | Denville | B1212-6S | |
| Nuclease-Free, Pure Water System | EMD Millipore | ||
| 5424 centrifuge | Eppendorf | 22621408 | |
| 5804R centrifuge | Eppendorf | 22623508 | Both 15 ml tube and plate rotators, preferably a centrifuge that can go up to 2,500 x g. |
| Safe-Lock Tube 1.5 mL, Natural | Eppendorf | 22431021 | |
| 5 mL Tube, DNA LoBind Tube | Eppendorf | 30108310 | |
| 5430R Centrifuge | Eppendorf | 022620645 | Any plate rotator centrifuge will work |
| Hybex Microsample Incubator | Fisher Scientific | 1057-30-0 | |
| Hybex 0.2 mL Tube Block | Fisher Scientific | 1057-31-0 | |
| TruSeq Amplicon – Cancer Panel | Illumina | FC-130-1008 | 96 reactions |
| TruSeq Custom Amplicon | Illumina | PE-940-1011 | 96 reactions |
| TruSeq Custom Amplicon Index Kit | Illumina | FC-130-1003 | 96 Indices, 384 Samples |
| MiSeq Reagent Kit v3, 500 Cycles | Illumina | MS-102-3003 | |
| MiSeq Reagent Kit v2, 300 Cycles | Illumina | MS-102-2002 | |
| MiSeq Reagent Kit v2, 500 Cycles | Illumina | MS-102-2003 | |
| Experiment Manager | Illumina | 1.3 or higher | |
| MiSeq Reporter | Illumina | 2.0 or higher | |
| Sequencing Analysis Viewer | Illumina | 1.8 or higher | |
| TruSeq Index Plate Fixture and Collar Kit | Illumina | FC-130-1007 | |
| MiSeq v2 | Illumina | SY-410-1003 | |
| TruSeq Custom Amplicon Filter Plate | Illumina | FC-130-1006 | |
| Index Adapter Replacement Caps | Illumina | 11294657 | |
| Qubit 2.0 | Invitrogen | Q32866 | |
| Qubit 0.5 ml Tubes | Invitrogen | Q32856 | |
| Qubit dsDNA Broad Range Assay Kit | Invitrogen | Q32853 | |
| DynaMa6-96 Magnetic Stand, Side Skirted | Invitrogen | 120.27 | |
| GeneAmp PCR System 9700 (gold/silver block) | Life Technologies | N8050200 | |
| Gentra Puregene Blood Kit | Qiagen | 158489 | |
| Deparaffinization Solution (16ml) | Qiagen | 19093 | |
| Buffer ATL (4x50ml) | Qiagen | 939011 | |
| Protein Precipitation Solution (50 ml) | Qiagen | 158910 | |
| DNA Hydration Solution (100ml) | Qiagen | 158914 | |
| Glycogen Solution (500 μl) | Qiagen | 158930 | |
| Qiagen Proteinase K | Qiagen | 19133 | |
| Rnase (5ml) | Qiagen | 158924 | |
| Nuclease-Free Water (10 x 50 ml) | Qiagen | 129114 | |
| Pestles | USA Scientific | 1415-5390 | |
| TipOne RPT 10 ul elongated filter pipet tips in sterilized racks, 10 racks of 96 tips (960 tips). | USA Scientific | 1180-3810 | |
| TipOne RPT 100 ul natural, beveled filter pipet tips in sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-1840 | |
| TipOne RPT 200 μl natural, beveled filter pipet tips in racks, sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-8810 | |
| TipOne RPT 20 μl natural, beveled filter pipet tips in racks, sterilized racks, 10 racks of 96 tips (960 tips) | USA Scientific | 1180-1810 | |
| TipOne RPT 1000 μl natural, graduated XL filter pipet tips in | USA Scientific | 1182-1830 |
References
- Gerlinger, M., et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 366 (10), 883-892 (2012).
- Campbell, P. J., et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 467 (7319), 1109-1113 (2010).
- Ding, L., et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 481 (7382), 506-509 (2012).
- Frampton, G. M., et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature Biotechnol. 31 (11), 1023-1031 (2013).
- Patel, J. P., et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 366 (12), 1079-1089 (2012).
- Forbes, S. A., et al. COSMIC (the Catalogue of Somatic Mutations in Cancer ): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 38 (Database Issue), 652-657 (2010).
- Shih, A. H., Abdel-wahab, O., Patel, J. P., Levine, R. L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 12 (9), 599-612 (2012).
- Liersch, R., Müller-Tidow, C., Berdel, W. E., Krug, U. Prognostic factors for acute myeloid leukaemia in adults – biological significance and clinical use. Br J Haematol. 165 (1), 17-38 (2014).
- Bacher, U., Schnittger, S., Haferlach, T. Molecular genetics in acute myeloid leukemia. Curr Opin Oncol. 22 (6), 646-655 (2010).
- Subramanian, J., Govindan, R. Lung cancer in "Never-smokers": a unique entity. Oncology (Williston Park). 24 (1), 29-35 (2010).
- Sakashita, S., Sakashita, M., Tsao, M. S. Genes and pathology of non-small cell lung carcinoma. Semin Oncol. 41 (1), 28-39 (2014).
- Arcila, M. E., Chaft, J. E., Nafa, K. Prevalence clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res. 18 (18), (2012).
- Gandhi, L., et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol. 32 (2), 68-75 (2014).
- Sheikhha, M. H., Awan, A., Tobal, K., Liu Yin, J. A. Prognostic significance of FLT3 ITD and D835 mutations in AML patients. Hematol J. 4 (1), 41-46 (2003).
- Mazières, J., et al. Lung cancer that harbors an HER2 mutation epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 31 (16), 1-8 (2014).
- Robinson, J. T., et al. Integrative Genomics Viewer. Nat Biotechnol. 29 (1), 495-500 (2011).
- Forbes, S. A., et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43 (Database issue), D805-D811 (2014).
- Daber, R., Sukhadia, S., Morrissette, J. J. Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genetics. 206 (12), 441-448 (2013).
- Haraksingh, R. R., et al. Genome-Wide Mapping of Copy Number Variation in Humans: Comparative Analysis of High Resolution Array Platforms. PLoS ONE. 6 (11), e27859 (2011).
- de Leeuw, N., et al. SNP Array Analysis in Constitutional and Cancer Genome Diagnostics – Copy Number Variants, Genotyping and Quality Control. Cytogenet Genome Res. 135 (3-4), 212-221 (2011).
