Generation of Genetically Modified Mice through the Microinjection of Oocytes
Summary
The microinjection of mouse oocytes is commonly used for both classic transgenesis (i.e., the random integration of transgenes) and CRISPR-mediated gene targeting. This protocol reviews the latest developments in microinjection, with a particular emphasis on quality control and genotyping strategies.
Abstract
The use of genetically modified mice has significantly contributed to studies on both physiological and pathological in vivo processes. The pronuclear injection of DNA expression constructs into fertilized oocytes remains the most commonly used technique to generate transgenic mice for overexpression. With the introduction of CRISPR technology for gene targeting, pronuclear injection into fertilized oocytes has been extended to the generation of both knockout and knockin mice. This work describes the preparation of DNA for injection and the generation of CRISPR guides for gene targeting, with a particular emphasis on quality control. The genotyping procedures required for the identification of potential founders are critical. Innovative genotyping strategies that take advantage of the “multiplexing” capabilities of CRISPR are presented herein. Surgical procedures are also outlined. Together, the steps of the protocol will allow for the generation of genetically modified mice and for the subsequent establishment of mouse colonies for a plethora of research fields, including immunology, neuroscience, cancer, physiology, development, and others.
Introduction
Animal models, both in vertebrates and invertebrates, have been instrumental to examining the pathophysiology of human conditions such as Alzheimer's disease1,2. They are also invaluable tools to search for disease modifiers and to ultimately develop novel treatment strategies in the hope of a cure. Although each model has intrinsic limitations, the use of animals as entire systemic models is vital to biomedical research. This is because the metabolic and complex physiological environment cannot be entirely simulated in tissue culture.
To date, the mouse remains the most common mammalian species used for genetic manipulation because it features several advantages. The physiological processes and genes associated with diseases are highly conserved between mice and humans. The mouse was the first mammal to have its full genome sequenced (2002), one year before the human genome (2003). Aside from this wealth of genetic information, the mouse has good breeding capacities, a fast development cycle (6 weeks from fertilization to weaning), and a reasonable size. All these advantages, coupled with physiological indicators, such as distinct coat colors (required for crossing strategies), made the mouse an attractive model for genetic manipulation. Notably, in the very early age of modern genetics, Gregor Mendel started working on mice before moving to plants3.
Gene transfer techniques resulted in the generation of the first transgenic mouse over three decades ago4, initially created using viral delivery. However, researchers soon realized that one of the main challenges of mouse transgenesis was the inability to control the fate of the exogenous DNA. Because the viral delivery of transgenes into mouse oocytes resulted in multiple copies integrated randomly into the genome, the possibility of establishing subsequent transgenic lines was limited.
One such limitation was overcome when Gordon et al. generated the first transgenic mouse line by microinjection5,6. This began the era of recombinant DNA technology, and the parameters influencing the outcome of a microinjection session have been widely studied7. Although microinjection does not allow for control over the integration site of the transgene (which eventually results in specific expression levels for each founder mouse), the main advantage of pronuclear microinjection remains the formation of concatemers (i.e., arrays of multiple copies of the transgene, linked in series) before genomic integration5. This characteristic has been used over the years to establish thousands of transgenic mouse lines that overexpress a gene of interest. Since then, transgenesis, the artificial modification of an organism's genome, has been extensively used to identify the role of single genes in the occurrence of diseases.
A further key achievement in manipulating the mouse genome was reached when Mario Capecchi successfully disrupted a single gene in the mouse, opening the era of gene targeting8. However, major drawbacks quickly emerged from ES cell-based gene targeting, including the challenges of culturing ES cells, the somewhat variable degree of chimerism, and the length of the process (i.e., 12-18 months, minimum, to obtain the mouse).
Recently, advances in new technologies, such as engineered endonucleases (e.g., zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN), and clustered regularly interspaced short palindromic repeats (CRISPR/Cas9)) have emerged as alternative methods to accelerate the process of gene targeting in mice9,10. These endonucleases can readily be injected into mouse oocytes by microinjection, allowing for the generation of gene-targeted mice in as little as 6 weeks.
Since the first report on the use of CRISPR for genome editing11, this bacterial adaptive immune system has superseded ZFN and TALEN because of its many advantages, including the ease of synthesis and the ability to target multiple loci at once (referred to as "multiplexing"). CRISPR was first used for gene targeting in mice12 and has since been applied to countless species, from plants to humans13,14. To date, there is no report of a single species resistant to CRISPR genome editing.
The two main limiting steps of the generation of transgenic mice are the injection of oocytes and the reimplantation of these oocytes into pseudo-pregnant females. Although this technique has been described by us15 and others16, recent technical improvements in mouse embryology and gene transfer techniques have revolutionized the process of generating genetically modified mice. These improvements will be described herein.
Protocol
All procedures have been approved by the University of New South Wales Animals Care and Ethics Committee.
1. Preparation of the Transgene (Random Integration)
- Analytical agarose gel electrophoresis.
- Digest the plasmid to excise the transgene using appropriate enzymes (1 h incubation) or fast-digest enzymes (15 to 30 min incubation) in a thermocycler following the manufacturer's recommendations (see Figure 2A and its legend).
- Cast a 1% tris-acetate-ethylenediaminetetraacetic acid (EDTA) (TEA) agarose gel, stained with 0.5-1.0 µg/mL ethidium bromide (EtBr).
NOTE: Use caution when using EtBr; it is a potent mutagen. Please use appropriate personal protective equipment (refer to the material safety data sheet). - Load a 1 kb molecular weight marker.
- Load the linearized fragments (i.e., transgene and backbone).
- Run the electrophoresis at 100 V for 45 min.
- Visualize the gel on an ultraviolet (UV) transilluminator to check that the digestion is complete and to confirm the correct size of the transgene (i.e., 7,338 bp; see Figure 2A).
- Preparative agarose gel electrophoresis.
- Cast a 1% TAE agarose gel stained with a low-toxicity gel stain. Load 8 aliquots (1 µg each) of linearized transgene without a molecular weight marker and run the electrophoresis at 100 V for 45 min. Use a scalpel to excise all 8 bands corresponding to the linearized transgene.
- Extract the DNA using a gel extraction kit by following the manufacturer's recommendations (see the Table of Materials).
- Melt the gel fragments at 50 °C in 1.5-mL tubes for at least 15 min. Precipitate it with isopropanol and then add the binding buffer.
- Combine the entire DNA by pipetting it all into one column. Elute into nuclease-free microinjection buffer (8 mM Tris-HCl and 0.15 mM EDTA). Filter through a 0.22 µm microcentrifuge filter and centrifuge for 60 s at 12,000 x g.
- Measure the concentration and the quality of the DNA using a spectrophotometer. Check that the A260/A280 ratio is around 1.8 (i.e., no contamination by proteins) and the A260/A230 ratio is at least 2.0 (i.e., no contamination by organic solvents). Dilute down to 3 ng/µL in nuclease-free microinjection buffer, aliquot (20-50 µL), and freeze at -20 °C.
2. Synthesis of CRISPR Components (Gene Targeting)
- Cas9 mRNA.
- Dilute the Cas9 mRNA (obtained from a commercial source, see the Table of Materials) to a concentration of 1 µg/µL in nuclease-free microinjection buffer.
- Aliquot in 2 µL and freeze at -80 °C.
- Single-guide RNA (SgRNA).
- Identify the desired two-target genomic sequences (guides) using a computational design tool allowing minimal potential off-target activity17.
- For gene knockout, systematically select two guides located a few hundred base-pairs (bp) apart, in opposite directions, and include the start codon of the gene of interest. For homology direct repair, select two overlapping guides (whenever possible), in opposite directions.
NOTE: As an example, the following guides have recently been successfully co-injected to disrupt the first exon of the TPM4.2 gene:
Guide 1: 5' CGCCATCCAGTTCGCGCTGC 3';
Guide 2: 5' CAGAACGATTGAGCTATGGC 3'
- For gene knockout, systematically select two guides located a few hundred base-pairs (bp) apart, in opposite directions, and include the start codon of the gene of interest. For homology direct repair, select two overlapping guides (whenever possible), in opposite directions.
- Order a set of primers as described in Table 1.
- Synthesize a linear DNA template.
- Dilute px33012 plasmid to 10 ng/µL in nuclease-free water.
- Prepare the master mix as indicated in Table 1. Add the polymerase at the end and keep the master mix on ice.
- Mix and split this into 8 PCR tubes (19 µL/tube). Add 1 µL of px330 per tube and run the PCR under the following conditions: 1 min at 98 °C; 40 cycles of 98 °C for 10 s, 64 °C for 30 s, and 72 °C for 15 s; and a final elongation at 72 °C for 5 min. Hold at 4 °C.
- Purify the PCR products using a PCR purification kit (see the Table of Materials), a manifold, and a vacuum source (for faster processing). Apply the maximum vacuum strength (i.e., 8 mbar).
- Add 5 volumes (100 µL) of binding buffer to 1 volume of the PCR sample and mix.
- Place a (provided) silica membrane spin column in a (provided) 2-mL collection tube for centrifugation, or on the manifold for vacuum processing. To bind DNA, successively apply all 8 samples to the column and vacuum or centrifuge for 60 s at 12,000 x g for each load.
- To wash, add 0.75 mL of wash buffer (buffer PE) to the column and vacuum or centrifuge for 60 s at 12,000 x g. Centrifuge the column for 60 s at 12,000 x g to eliminate residual ethanol.
- Place the column in a clean 1.5-mL microcentrifuge tube. To elute the DNA, add 30 µL of nuclease-free water to the center of the membrane, let the column stand for 1 min, and centrifuge for 60 s at 12,000 g.
- Measure the concentration using a spectrophotometer; typically, the concentration should be 100 ng/µL or higher. Check that the A260/A280 ratio is around 1.8 (i.e., no contamination by proteins) and the A260/A230 ratio is at least 2.0 (i.e., no contamination by organic solvents).
- In vitro transcription using a T7 RNA synthesis kit.
- Prepare the master mix, as indicated in Table 1, and incubate for 3 h at 37 °C in a thermocycler. Add 28 µL of nuclease-free water and 2 µL of DNase I and incubate for another 15 min at 37 °C in a thermocycler.
- RNA purification using spin columns.
- Dissolve the powder contained in the provided spin column in 650 µL of nuclease-free microinjection buffer, carefully removing all air bubbles. Cap the tube and hydrate for 5 – 15 min at room temperature.
- Remove the blue cap at the bottom and place the column in a 2-mL tube. Centrifuge for 2 min at 750 x g and room temperature. Place the column in a fresh 1.5-mL tube and apply 50 µL of the RNA solution dropwise to the center, without touching the column wall. Spin the column for 2 min at 750 x g.
- Measure the RNA concentration using a spectrophotometer (the typical yield of one reaction is 30 – 50 µg). Check that the A260/A280 ratio is around 2.0 (i.e., no contamination by proteins) and the A260/A230 ratio is at least 2.0 (i.e., no contamination by organic solvents). Store the sgRNAs at -80 °C until use.
- Assess the quality of the RNA using the 1% TAE gels routinely used for DNA electrophoresis. Run 200-400 ng of the DNA template and the corresponding sgRNA in parallel. The RNA band (≈ 100 bp) should be slightly bigger than the DNA band (see Figure 3A).
- Identify the desired two-target genomic sequences (guides) using a computational design tool allowing minimal potential off-target activity17.
3. Donor Template
- Order commercially synthesized single-stranded oligonucleotides (ssOligos, see the Table of Materials) for point mutations or for the integration of small sequences up to 50 bp; homology arms are typically 60-90 bp long.
- For larger insertions, use a donor plasmid as a template. Generate a suitable plasmid using either the classic cloning method or de novo synthesis from a commercial source.
NOTE: A minimum of 800 bp per homology arm is recommended. The size of the backbone has no influence on the efficiency of homology direct repair (HDR).
4. Injection Mix
- Dilute the Cas9 mRNA to 50 ng/µL and the sgRNAs to 12.5 ng/µL (25 ng/µL in total) using nuclease-free microinjection buffer for gene knockout experiments. Add the donor template (ssOligo or plasmid) at a concentration of 200 ng/µL for genome editing experiments based on homology direct repair.
NOTE: Dilution volumes can be easily determined using online tools, such as a resuspension calculator18. - Keep each aliquot on ice during the microinjection session and discard afterwards (do not re-freeze the injection mix).
5. Scrotal Vasectomy
NOTE: Two types of vasectomy are commonly performed in mice: abdominal and scrotal. The latter is less invasive and has been previously described15.
- Autoclave all stainless steel surgical instruments.
- Determine the weight of the mouse using a weigh scale and anesthetize the males by intraperitoneal (i.p.) injection with injectable anesthetics (ketamine 100 mg/kg, xylazine 10 mg/kg).
- Monitor the loss of toe pinch reflex and once the mouse is sedated place it on top of a heating pad.
- Disinfect the skin around the testes with alternating wipes of a topical antiseptic such as chlorhexidine and 70% ethanol.
- Push the testes into the scrotal sac and make a skin incision with a scalpel.
- Visualize the testis, cauda epididymis, and vas deferens.
- Grab the vas deferens with forceps, hold it, and cauterize on each side of the forceps to remove 3 mm of the vas deferens.
- Perform the same procedure on the second testis.
- Close the skin incisions with wound clips and monitor the mouse closely until full recovery.
6. Superovulation (Oocytes Donors) and Time-mating (Pseudo-pregnant Females)
NOTE: The technique to generate a suitable number of fertilized oocytes and plugged foster females for reimplantation has been described elsewhere15.
- Inject 10 females i.p. with 5 IU of pregnant mare's serum gonadotropin (PMSG; in 100 µL) at around 12 pm on day 1.
- Inject the females i.p. with 5 IU of human chorionic gonadotropin (hCG; in 100 µL) 46-48 h later (at around 11 am on day 3).
- Immediately mate the females with single-housed males overnight.
7. Pronuclear (random-integration) and Cytoplasmic (Gene-targeting) Injections
- Cull the superovulated mice by cervical dislocation, expose the abdomen, and access the ovaries and oviducts, as previously described15. Dissect the oviducts and harvest the cumulus-oocyte-complexes (COCs) under a stereomicorscope15. Place them in a drop of potassium simplex optimization medium supplemented with amino acids (KSOMaa) following the traditional method15.
- Purify the oocytes by adding approximatively 1 µL of hyaluronidase (10 mg/mL) to the drop of KSOMaa medium using an aspirator mouth piece and place the dish in a 37 °C / 5% CO2 incubator for 30 s to 1 min.
- Wash the oocytes with 4 fresh drops of KSOMaa medium using the aspirator mouth piece and transfer them to a final drop of KSOMaa medium overlaid with mineral oil (around 2 mL). Place the dish in the 37 °C / 5% CO2 incubator until the oocytes are ready for injection.
- Pronuclear injection (random integration).
- Visualize the eggs under the stereoscopic microscope and transfer approximately 50 eggs to the injection chamber (a drop of M2 medium overlaid with mineral oil) using the aspirator mouth piece.
- Transfer the injection chamber to the inverted microscope and inject the fertilized oocytes (two visible pronuclei) with a few picoliters of the injection mix, targeting a pronucleus (any of the pronuclei can be targeted). Visualize a successful injection by observing the swelling of the pronucleus.
- Cytoplasmic injection (gene targeting).
- Transfer approximately 50 eggs to a drop of KSOMaa containing 5 µg/mL Cytochalasin B using the mouth piece and incubate for 5 min in the 37 °C / 5% CO2 incubator.
- Transfer the eggs to the injection chamber using the mouth piece.
- Inject few picoliters of the injection mix into the cytoplasm at very low pressure (50-100 hPa), using the compensation pressure of the automated microinjector where possible.
- After injection, transfer the oocytes back into a drop of KSOMaa (using the mouth piece) and keep them in the 37 °C/5% CO2 incubator, until loaded for reimplantation into the oviduct of pseudo-pregnant females.
8. Reimplantation
- Autoclave all stainless steel surgical instruments.
- Determine the weight of the mouse using the weigh scale and anesthetize the females by intraperitoneal (i.p.) injection with injectable anesthetics (ketamine 100 mg/kg, xylazine 10 mg/kg).
- Monitor the loss of toe pinch reflex and once the mouse is sedated place it on top of a heating pad.
- Clip a large area of the fur around the dorsal midline of the female mouse.
- Disinfect the exposed skin with alternating wipes of a topical antiseptic such as chlorhexidine and 70% ethanol.
- Expose the reproductive tract following traditional method15. Make a 1 cm long skin incision parallel to the dorsal midline, cut the muscle and grab the fat pad with a forceps, then gently pull the ovary out until its attached oviduct and uterus are clearly visible.
- Fix the fat pad with a vessel clamp. Visualize the oviduct under the stereoscopic microscope and using a pair of micro-scissors make an incision into the wall of the oviduct few millimeters upstream of the ampulla.
- Under the stereoscopic microscope, load 25 microinjected eggs into the glass capillary connected to the mouth piece (the internal diameter of the capillary should be around 120 µm wide). Introduce the glass capillary into the oviduct and expel until an air bubble is visible inside the ampulla.
- Gently remove the glass capillary and place the reproductive tract back into the abdomen. Suture the incision with 3-0 non-absorbable surgical sutures, then close with wound clips. Monitor the mouse closely until full recovery.
9. Genotyping Strategies/Sequencing
NOTE: Isolate the genomic DNA from 2-mm tail or ear biopsies, following relevant animal ethics regulations.
- Fast genomic DNA extraction (random integration).
- Lyse the tissue samples (~2 mm) at 95 °C for 1 h in 100 µL of alkaline lysis reagent (25 mM sodium hydroxide and 0.2 mM EDTA, pH = 12).
- Add 100 µL of neutralizing reagent (40 mM Tris-HCl, pH = 5).
- Centrifuge for 5 min at 12,000 g and 4 °C.
- High-quality genomic DNA extraction (gene targeting).
- Add 500 µL of tail buffer (50 mM Tris, pH = 8; 100 mM EDTA, pH = 8; 100 mM NaCl; 1% SDS, and 0.5 mg/mL proteinase K, freshly added).
- Incubate overnight at 55 °C.
- Mix for 5 min by inverting (do not vortex).
- Add 250 µL of saturated NaCl (6 M) and mix for 5 min by inverting (do not vortex).
- Spin for 5-10 min at 12,000 x g and 4 °C and pour the supernatant into new tube.
- Add 500 µL of isopropanol and mix for 5 min by inverting (do not vortex).
- Spin for 10 min at 12,000 x g and room temperature.
- Decant the supernatant (the pellet is invisible and sticks to the tube).
- Wash with 1 mL of 70% ethanol.
- Spin for 5-10 min at 12,000 x g and room temperature. Remove the supernatant with care, as the white pellet is not sticky anymore and can be lost.
- Air dry this for ~1 h.
- Add 400 µL of TE buffer (10 mM Tris and 1 mM EDTA, pH = 8).
- Dissolve the DNA at 55-60 °C for 2 h.
- Primer design.
- Design primers a minimum of 20 bp long using a computational tool19.
NOTE: For random integration, the primers should hybridize with the transgene, generating a distinct fragment of 200-800 bp. For gene targeting, design the primers so that they hybridize with the genomic DNA, generating a distinct fragment a few hundred bp around the cut sites. For large insertions, the primers can sit on the transgene, but confirmation of the targeted integration should subsequently be performed using primer walking or a similar technique.
- Design primers a minimum of 20 bp long using a computational tool19.
- PCR genotyping.
- For each genomic modification, design a new PCR protocol and test it empirically. Consider the length of the generated fragment and the melting temperature (Tm) of each pair of primers.
- Sequencing.
- For gene targeting, send PCR products to a Sanger sequencing service provider.
Representative Results
Below, the workflows for microinjection in the case of random integration and CRISPR-mediated gene targeting are described (Figure 1).
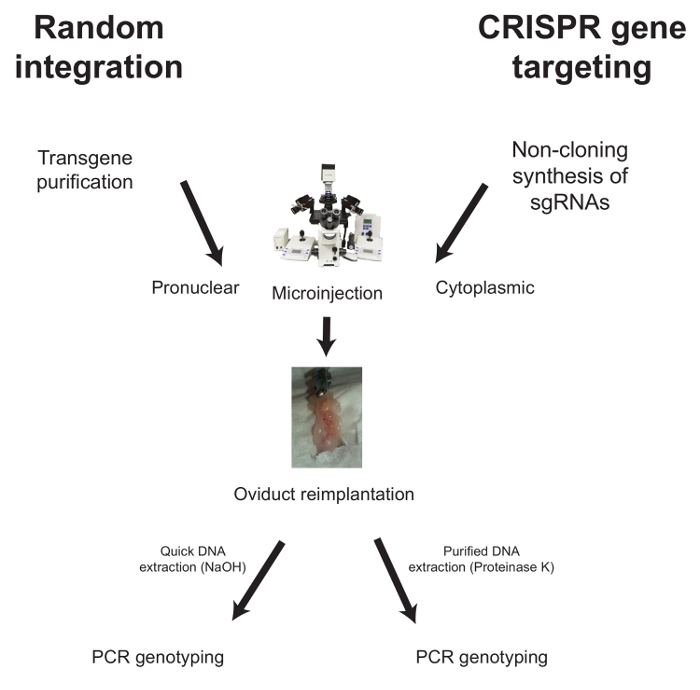
Figure 1: Typical Workflow for the Generation of Genetically Modified Mice. For random integration, the purified transgene is injected into the pronucleus of fertilized oocytes before oviduct transfer into plugged foster females. Analysis of the progeny is done by PCR after a quick genomic DNA extraction. For CRISPR gene targeting, sgRNAs are synthesized using the non-cloning method described here, and two guides are co-injected (together with Cas9 mRNA) into the cytoplasm of fertilized oocytes. This strategy is used for the subsequent PCR-based analysis of the progeny, which requires highly purified genomic DNA. Please click here to view a larger version of this figure.
Although the microinjection of oocytes and the reimplantation of these injected eggs are the two main limiting steps that require technical skills and training, the quality of the injection mix is of the utmost importance to successfully generate genetically modified mice. The quality of the transgene purification (Figure 2A) and the sgRNAs synthesis (Figure 3A) should be systematically assessed on analytical agarose gels. Potential contaminations of the mix should also be checked when measuring the concentrations with a spectrophotometer, as the different values of absorption directly depend upon the degree of purity of the solution (see steps 1.2.3, 2.2.3.5, and 2.2.5.3).
Likewise, the identification of potential founder mice heavily relies upon innovative genotyping strategies. This work illustrates a typical readout of a microinjection session, both for random integration (Figure 2B) and for gene targeting (Figure 3B). The DNA extraction protocols are also different depending upon the type of experiments; subsequent PCRs rely on primers that hybridize either on the transgene or on the genomic DNA, respectively. The strategies presented here allow for the easy troubleshooting and streamlining of each step required to produce genetically modified mice.
Although the quantitative outcome of a microinjection session varies depending upon the experience of the experimenter, once each step of the protocol is optimized, the percentage of transgenic pups obtained should typically reach 10-25% for random integration, as illustrated in Figure 2B (4 founders out of 15 pups born = 26.6%). This somewhat "low" efficiency contrasts with the reliable ability of the CRISPR components to edit the mouse genome. Indeed, the number of founders generated is generally higher when using CRISPR for gene targeting and ranges from 25-100% (see Figure 3B; 3 founders out of 13 pups = 23%). It is not uncommon to obtain an entire progeny that is successfully homozygous when they are edited using CRISPR.
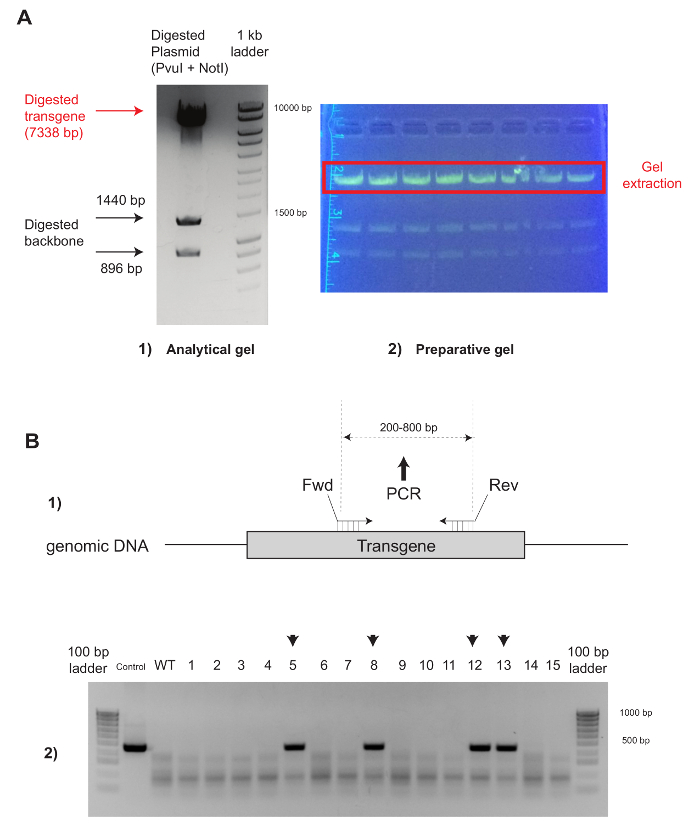
Figure 2: Quality Control and Genotyping Strategy for the Successful Production of Transgenic Mice for Overexpression. (A) The plasmid is digested for 1 h with PvuI and NotI. Complete digestion and correct sizes (i.e., transgene = 7,338 bp, backbone = 1,440 + 896 bp) are checked on an analytical gel (1). The extraction of several digestion products from the preparative gel (2) generates an increased concentration of the transgene. Prolonged exposure to UV should be avoided, and the use of a blue light in lieu of a UV transilluminator is recommended. (B) Genotyping strategy (1): The primers should sit on the transgene and should be designed to generate a fragment of 200 – 800 bp. When genotyping is performed (2), it is recommended to include a positive control (i.e., the injection mix used for microinjection) and a negative control (i.e., genomic DNA from a WT mouse). Please click here to view a larger version of this figure.
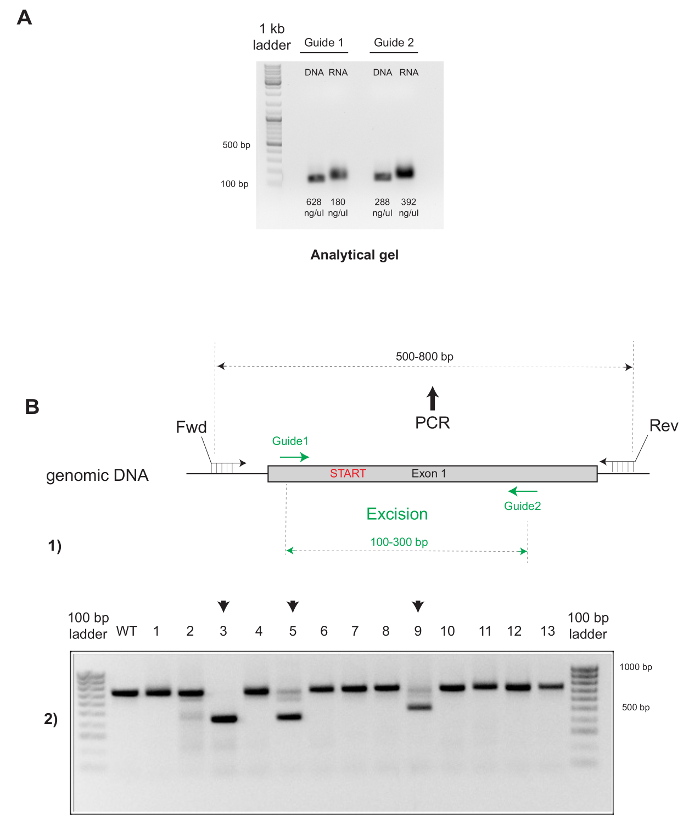
Figure 3: Quality Control and Genotyping Strategy for the Successful Production of Gene-targeted (KO) Mice. (A) The quality of the sgRNAs produced by in vitro transcription is assessed by running the DNA template and the generated RNA in parallel for each guide. The size of the RNA is slightly bigger than the DNA. Should the quality be satisfactory (i.e., no smear bands) the concentrations are measured. (B) (1) Design of the two guides (opposite directions) encompassing the Start codon in Exon 1. The primers are selected to hybridize with the genomic DNA outside of the region eventually excised by the two guides. Typically, this strategy allows for direct identification of heterozygous (# 5 and 9) and homozygous (# 3) KO founders using PCR (2). PCR products of homozygous animals do not display any WT band. Please click here to view a larger version of this figure.
| Reaction | Components | Volume | Note |
| Synthesis of linear DNA template (2.5) |
Nuclease free water | 97.2 µL | |
| 5x HF buffer | 36 µL | ||
| MgCl2 | 9 µL | ||
| 10 mM dNTPs | 9 µL | ||
| 10 µM fwd primer | 9 µL | 5’TTAATACGACTCACTATAGN20 gttttagagctagaaatagcaagttaaaata aggctagtc 3’ |
|
| 10 µM rev primer | 9 µL | 5’ AAAAGCACCGACTCGGTGCC 3’ | |
| Proof-reading polymerase | 1.8 µL | ||
| IVT using a T7 RNA synthesis kit (2.6) |
Nuclease free water | X µL | Make up to 20 µL total volume |
| NTP buffer mix | 10 µL | ||
| Template DNA | ≈ 300 ng | ||
| T7 RNA polymerase | 2 µL |
Table 1: Composition of the Different Master Mixes Used in this Study. Synthesis of the linear DNA template: The T7 promoter minimal sequence (TTAATACGACTCACTATAG) is upstream of the 20-bp sequence (guide; N20 identified using the CRISPR design tool) and a sequence complementary to the expression vector (gttttagagctagaaatagcaagttaaaataaggctagtc). In vitro transcription (IVT): Depending upon the concentration of the DNA template, the final volume should be adjusted to 20 µL with nuclease-free water.
Discussion
Critical steps within the protocol
The generation of genetically modified mice is known to be technically challenging. However, the protocol presented here is an optimized and simplified method that allows one to master and troubleshoot the technique in record time. There are two steps necessary for the successful completion of the technique. First, the synthesis of linear DNA templates (for the synthesis of sgRNAs) can be achieved without magnesium chloride (MgCl2). However, it is highly recommended to systematically add MgCl2 to the master mix because the partial hybridization of the forward primer is often prevented in the absence of MgCl2. In addition, it is critical for the targeted genomic sequence to not be present in any part of the donor template (in the case of targeted integration), otherwise the Cas9 nuclease will cut it, preventing the occurrence of HDR.
Modifications and troubleshooting
Although this protocol for generating genetically modified mice has been optimized and streamlined over the years, there are some considerations relating to the yield of good quality oocytes for microinjection and the plugging rate of foster females. The choice of the background strain is critical in regard to the number of live pups resulting from a microinjection session. C57BL/6 is the most commonly used background for mouse models of diseases. However, it is also one of the less efficient backgrounds in terms of resistance to microinjection20. Furthermore, the age of the female is also critical to yield a large number of oocytes; it is recommended to avoid mice 5-8 weeks of age, regardless of the background, as this is the least favorable period of response to hormonal stimulation21. In our experience, 3-to 4-week-old C57BL/6 mice reliably yield 20-30 eggs per female. It is important to note that a new technique (referred to as ultra-superovulation) has produced up to 100 eggs per female and has recently been used to generate oocytes for microinjection22. However, the use of ultra-superovulation for microinjection is generally hampered by the need to artificially fertilize (in vitro fertilization) such a large number of eggs.
To obtain plugged foster females suitable for reimplantation, female mice are mated with vasectomized males (the background of these mice is not critical). To increase the number of plugged recipients, careful examination of the stage of estrus and the selection of suitable females can be performed23. Synchronization of the females' cycles can also be induced by exposing the females to vasectomized males two days prior to mating (the so-called "Whitten effect")24.
Limitations of the technique
Once optimized, the procedure should reliably allow one to systematically generate genetically modified mice during each microinjection session. Nonetheless, there are two main limitations to the procedure that correlate with the size of the attempted genomic modification.
Very small genomic modifications, such as point mutations, are relatively easy to generate, but they are difficult to identify. When genotyping is performed, the PCR products are directly sequenced by Sanger sequencing. However, the direct microinjection of CRISPR components into mouse oocytes often generates mosaic animals because the Cas9 remains active for quite some time and different genomic modifications can occur after the first division of the oocyte. This confounding effect complicates the identification of discrete mutations among various alleles, and next-generation sequencing (NGS) can help with challenging readouts. Sensitive techniques, such as tetra-primer ARMS-PCR,25 can also help with genotyping the colony after the identification of the founders by sequencing.
Conversely, inserting large pieces of DNA in a targeted fashion remains challenging. The larger the exogenous DNA, the lesser the efficiency of HDR. Consequently, new strategies, such as the use of long ssOligos donors (in lieu of plasmids)26 or HDR-independent targeted integration27, are currently being developed.
Significance of the protocol
This protocol presents significant advances because the four main steps (i.e., preparation of the transgene or CRISPR components, microinjection, reimplantation, and genotyping) have been optimized, tested, and validated in our laboratory.
The random integration of transgenes is widely used for overexpression studies for two main reasons. First, to avoid the potential "position effect," the targeted integration of a transgene using CRISPR in "safe harbor" loci, such as the Rosa2628 and the Col1a29 loci, remains notoriously challenging, since the efficiency of HDR is low. Strategies to overcome this problem are forthcoming26,27, but random integration has so far proved more efficient. Besides, the generation of multiple transgenic lines overexpressing the same transgene with different levels of expression is of interest in studies involving treatment strategies to reverse a phenotype30. Plasmid-based transgenes are generated by mean of cloning, using ad hoc restriction enzymes to assemble essential fragments for the expression of a protein of interest. Generally, a transgene is made of at least three elements: a suitable promoter (enhancer regions are sometimes added); the coding sequence (cDNA), with or without intronic regions; and a PolyA tail to stabilize mRNA expression31.
Once assembled, the transgene is inserted into a plasmid containing bacterial replication elements and expanded in culture after transformation (typically heat-shock based). The purification method of the transgenes for microinjection is critical. This work describes the use of new-generation (low-toxicity) DNA stains for the more accurate visualization of the fragment of interest on the gel (compared to traditional crystal violet) and of a low mutagenic exposure (compared to ethidium bromide).
The non-cloning method described herein for gene targeting is very fast and allows for the synthesis of several sgRNAs in only 5-6 h. Only four steps are required: the identification of the targeted sequence, the synthesis of a linear DNA template containing a T7 promoter and the chosen target sequence, the synthesis of the sgRNA by in vitro transcription (IVT), and the purification of the sgRNA using spin columns.
In the early age of CRISPR mouse genome editing, the potential off-target effect was systematically assessed, although it has been showed to be very limited in mouse embryos32 and can easily be diluted out by mean of a breeding scheme wherein selected mice carry only the desired genomic modification. On-target activity was also systematically pre-assessed in vitro, using different guides and selecting the more active one(s)12. This practice is now less common, for several reasons. First, the on-target activity is assessed using transfection of immortalized mouse cell lines (typically N2a or NIH3T3) with CRISPR-encoding plasmids. This is a different method than the injection of CRISPR RNA components into mouse oocytes. Therefore, the results found in vitro do not always translate equally to oocytes. Furthermore, high-throughput experiments showed that most guides are active33. Consequently, on-target activity is not assessed, and thus far, there has been no evidence of a guide showing no activity in mouse oocytes. Together with the non-cloning method to synthesize sgRNAs, presented here, this greatly simplifies and streamlines the workflow, and multiple active guides can seamlessly be synthesized in one day.
CRISPR is considered a "disruptive technology," which is an innovation that changes the way techniques are performed. When microinjection was established, Brinster et al. showed that cytoplasmic transgenesis, although achievable, remained mostly inefficient7. Consequently, pronuclear microinjection remained the only common practice for almost 30 years, until the first demonstration of a successful CRISPR gene targeting in mice. This study showed that cytoplasmic microinjection could generate a similar or superior outcome compared with pronuclear delivery12. Obviously, because CRISPR components are typically made of RNA, it is evident that the cytoplasm is targeted. However, even when injected into the cytoplasm, donor templates can also successfully induce HDR. A high cytoplasmic concentration of templates enables their diffusion into the nucleus. Moreover, the use of piezo-driven cytoplasmic microinjection16 is not a requirement. A short incubation of the oocytes with a cytoskeleton inhibitor, such as Cytochalasin B, increases the survival of the oocytes34,35, making it sufficient to perform cytoplasmic injections without a piezo impact drive system. Likewise, the use of an improved culture medium, such as KSOMaa, decreases the blockage at the two-cell stage and improves the developmental capacities of the embryos compared to M16 medium36.
One-cell or two-cell (when cultivated overnight) embryos can be transferred to the oviduct. The conventional procedure is quite invasive, as it requires tearing the bursa that protects the ovary. It is also technically challenging, because the glass capillary needs to be inserted in the infundibulum15. The method presented here is much easier to learn, does not induce potential bleeding, and preserves the integrity of the ovary. It is based on work developed at Kumamoto University37.
Genotyping strategies and sequencing techniques are critical to identify potential founders. For random integration, the extraction of genomic DNA is based on a simple method adapted from Truett et al.38. Such a quick extraction method is sufficient to identify potential founders, since the primers are designed to hybridize with the transgene (often integrated as multiple copies). Conversely, gene targeting requires highly purified genomic DNA, since the PCR product encompasses the modified genomic region.
For gene knockout, the traditional method of injecting a single sgRNA generates small deletions (indels) or insertions, eventually knocking out the gene of interest12. These small modifications are difficult to identify and often require time-consuming, ligase-independent cloning and sequencing to confirm the nature of these mutations.
In this protocol, we took advantage of CRISPR multiplexing to develop the systematic co-injection of two sgRNAs encompassing the start codon of a gene of interest. This not only increases the likeliness of a gene knockout, but also the excision of a chunk of genomic DNA of a predefined size (100-300 bp), allowing for the easy identification of the founders by simple PCR. Of note, pups that do not carry the expected genomic excision could still be analyzed for small frameshift mutations, when only one sgRNA proved active. Likewise, in the case of gene editing, the systematic co-injection of two overlapping sgRNAs in opposite directions should increase the efficiency of HDR, since the cell repair mechanisms preferentially use one of the two strands39.
Future applications
The present protocol takes advantage of the latest developments in mouse embryology and gene targeting40 to simplify and streamline the process of generating various sophisticated mouse models of human conditions. These models play a pivotal role in helping to develop our understanding of pathomechanisms, ultimately aiding in the development of therapeutic strategies2. The optimization and streamlining of the reagents required for gene targeting or random integration are widely and readily applicable to any other mammalian species, such as rats, rabbits, and even to large animals like cattle.
Divulgations
The authors have nothing to disclose.
Acknowledgements
The authors thank the staff of the animal facility (BRC) for their ongoing support. This work was funded by the National Health and Medical Research Council and the Australian Research Council.
Materials
| Micropipette 0.1-2.5 ul | Eppendorf | 4920000016 | |
| Micropipette 2-20 ul | Eppendorf | 4920000040 | |
| Micropipette 20-200 ul | Eppendorf | 4920000067 | |
| Micropipette 100-1000 ul | Eppendorf | 4920000083 | |
| Molecular weight marker | Bioline | BIO-33025 | HyperLadder 1kb |
| Molecular weight marker | Bioline | BIO-33056 | HyperLadder 100 bp |
| Agarose | Bioline | BIO-41025 | |
| EDTA buffer | Sigma-Aldrich | 93296 | 10x – Dilute to 1x |
| Ethidium bromide | Thermo Fisher Scientific | 15585011 | |
| SYBR Safe gel stain | Invitrogen | S33102 | |
| Gel extraction kit | Qiagen | 28706 | |
| PCR purification kit (Qiaquick) | Qiagen | 28106 | |
| Vacuum system (Manifold) | Promega | A7231 | |
| Nuclease-free microinjection buffer | Millipore | MR-095-10F | |
| Ultrafree-MC microcentrifuge filter | Millipore | UFC30GV00 | |
| Cas9 mRNA | Sigma-Aldrich | CAS9MRNA | |
| CRISPR expressing plasmid (px330) | Addgene | 42230 | |
| Nuclease free water | Sigma-Aldrich | W4502 | |
| Phusion polymerase | New England Biolabs | M0530L | |
| T7 Quick High Yield RNA kit | New England Biolabs | E2050S | |
| RNA purification spin columns (NucAway) | Thermo Fisher Scientific | AM10070 | |
| ssOligos | Sigma-Aldrich | OLIGO STANDARD | |
| Donor plasmid | Thermo Fisher Scientific | GeneArt | |
| Hyaluronidase | Sigma-Aldrich | H3884 | |
| KSOMaa embryo culture medium | Zenith Biotech | ZEKS-100 | |
| Mineral oil | Zenith Biotech | ZSCO-100 | |
| M2 Medium | Sigma-Aldrich | M7167 | |
| Cytochalasin B | Sigma-Aldrich | C6762 | |
| Mouthpiece | Sigma-Aldrich | A5177 | |
| Glass microcapillaries | Sutter Instrument | BF100-78-10 | |
| Proteinase K | Applichem | A3830.0100 | |
| Dumont #5 forceps | Fine Science Tools | 91150-20 | |
| Iris scissors | Fine Science Tools | 91460-11 | |
| Vessel clamp | Fine Science Tools | 18374-43 | |
| Wound clips | Fine Science Tools | 12040-01 | |
| Clips applier | Fine Science Tools | 12018-12 | |
| Micro-scissors | Fine Science Tools | 15000-03 | |
| Cauterizer | Fine Science Tools | 18000-00 | |
| Non-absorbable surgical sutures (Ethilon 3-0) | Ethicon | 1691H | |
| 5% CO2 incubator | MG Scientific | Galaxy 14S | |
| Spectrophotometer | Thermo Fisher Scientific | Nanodrop 2000c | |
| Thermocycler | Eppendorf | 6321 000.515 | |
| Electrophoresis set up | BioRad | 1640300 | |
| UV Transilluminator | BioRad | 1708110EDU | |
| Thermocycler | Eppendorf | 6334000069 | |
| Stereoscopic microscope | Olympus | SZX7 | |
| Inverted microscope | Olympus | IX71 | |
| 2x Micromanipulators | Eppendorf | 5188000.012 | |
| Oocytes manipulator | Eppendorf | 5176000.025 | |
| Microinjector (Femtojet) | Eppendorf | 5247000.013 | |
| Mice C57BL/6J strain | Australian BioResources | C57BL/6JAusb |
References
- Ittner, L. M. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 142 (3), 387-397 (2010).
- Ittner, A. Site-specific phosphorylation of tau inhibits amyloid-beta toxicity in Alzheimer’s mice. Science. 354 (6314), 904-908 (2016).
- Marantz Henig, R. . The Monk in the Garden. , (2000).
- Jaenisch, R., Mintz, B. Simian virus 40 DNA sequences in DNA of healthy adult mice derived from preimplantation blastocysts injected with viral DNA. Proc Natl Acad Sci U S A. 71 (4), 1250-1254 (1974).
- Gordon, J. W., Ruddle, F. H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science. 214 (4526), 1244-1246 (1981).
- Gordon, J. W., Scangos, G. A., Plotkin, D. J., Barbosa, J. A., Ruddle, F. H. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc Natl Acad Sci U S A. 77 (12), 7380-7384 (1980).
- Brinster, R. L., Chen, H. Y., Trumbauer, M. E., Yagle, M. K., Palmiter, R. D. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc Natl Acad Sci U S A. 82 (13), 4438-4442 (1985).
- Capecchi, M. R. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 6 (6), 507-512 (2005).
- Carbery, I. D. Targeted genome modification in mice using zinc-finger nucleases. Génétique. 186 (2), 451-459 (2010).
- Sung, Y. H. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol. 31 (1), 23-24 (2013).
- Jinek, M. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Wang, H. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153 (4), 910-918 (2013).
- Kang, X. Introducing precise genetic modifications into human 3PN embryos by CRISPR/Cas-mediated genome editing. J Assist Reprod Genet. 33 (5), 581-588 (2016).
- Liang, P. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 6 (5), 363-372 (2015).
- Ittner, L. M., Götz, J. Pronuclear injection for the production of transgenic mice. Nat Protoc. 2 (5), 1206-1215 (2007).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc. 9 (8), 1956-1968 (2014).
- . Resuspension Calculator Available from: https://sg.idtdna.com/calc/resuspension/ (2017)
- Auerbach, A. B. Strain-dependent differences in the efficiency of transgenic mouse production. Transgenic Res. 12 (1), 59-69 (2003).
- Merriman, J. A., Jennings, P. C., McLaughlin, E. A., Jones, K. T. Effect of aging on superovulation efficiency, aneuploidy rates, and sister chromatid cohesion in mice aged up to 15 months. Biol Reprod. 86 (2), 49 (2012).
- Nakagawa, Y., et al. Ultra-superovulation for the CRISPR-Cas9-mediated production of gene-knockout, single-amino-acid-substituted, and floxed mice. Biol Open. 5 (8), 1142-1148 (2016).
- Byers, S. L., Wiles, M. V., Dunn, S. L., Taft, R. A. Mouse estrous cycle identification tool and images. PLoS One. 7 (4), e35538 (2012).
- Whitten, W. K. Modification of the oestrous cycle of the mouse by external stimuli associated with the male. J Endocrinol. 13 (4), 399-404 (1956).
- Ye, S., Dhillon, S., Ke, X., Collins, A. R., Day, I. N. An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res. 29 (17), E88 (2001).
- Yoshimi, K., et al. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun. 7, 10431 (2016).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. , (2016).
- Soriano, P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 21, 70-71 (1999).
- Liu, X., et al. A targeted mutation at the known collagenase cleavage site in mouse type I collagen impairs tissue remodeling. J Cell Biol. 130 (1), 227-237 (1995).
- Ke, Y. D. Short-term suppression of A315T mutant human TDP-43 expression improves functional deficits in a novel inducible transgenic mouse model of FTLD-TDP and ALS. Acta Neuropathol. 130 (5), 661-678 (2015).
- Auerbach, A. B. Production of functional transgenic mice by DNA pronuclear microinjection. Acta Biochim Pol. 51 (1), 9-31 (2004).
- Iyer, V., et al. Off-target mutations are rare in Cas9-modified mice. Nat Methods. 12 (6), 479 (2015).
- Zhou, Y. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 509 (7501), 487-491 (2014).
- Brinster, R. L., Chen, H. Y., Trumbauer, M. E., Avarbock, M. R. Translation of globin messenger RNA by the mouse ovum. Nature. 283 (5746), 499-501 (1980).
- Pinkert, C. A., Irwin, M. H., Johnson, L. W., Moffatt, R. J. Mitochondria transfer into mouse ova by microinjection. Transgenic Res. 6 (6), 379-383 (1997).
- Biggers, J. D., Summers, M. C. Choosing a culture medium: making informed choices. Fertil Steril. 90 (3), 473-483 (2008).
- Nakagata, N. Embryo transfer through the wall of the fallopian tube in mice. Jikken Dobutsu. 41 (3), 387-388 (1992).
- Truett, G. E. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques. 29 (1), 52-54 (2000).
- Richardson, C. D., Ray, G., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 34 (3), 339-344 (2016).
- Delerue, F., Ittner, L. M. Genome Editing in Mice Using CRISPR/Cas9: Achievements and Prospects. Clon. Transgen. 4, (2015).
