Culturing In Vivo-like Murine Astrocytes Using the Fast, Simple, and Inexpensive AWESAM Protocol
Summary
The AWESAM protocol described here is optimal for culturing murine astrocytes in isolation from other brain cells in a fast, simple, and inexpensive manner. AWESAM astrocytes exhibit spontaneous Ca2+ signaling, morphology, and gene expression profiles similar to astrocytes in vivo.
Abstract
The AWESAM (a low-cost easy stellate astrocyte method) protocol entails a fast, simple, and inexpensive way to generate large quantities of in vivo-like mouse and rat astrocyte monocultures: Brain cells can be isolated from different brain regions, and after a week of cell culture, non-astrocytic cells are shaken off by placing the culture dishes on a shaker for 6 h in the incubator. The remaining astrocytes are then passaged into new plates with an astrocyte-specific medium (termed NB+H). NB+H contains low concentrations of heparin-binding EGF-like growth factor (HBEGF), which is used in place of serum in medium. After growing in NB+H, AWESAM astrocytes have a stellate morphology and feature fine processes. Moreover, these astrocytes have more in vivo-like gene expression than astrocytes generated by previously published methods. Ca2+ imaging, vesicle dynamics, and other events close to the membrane can thus be studied in the fine astrocytic processes in vitro, e.g., using live cell confocal or TIRF microscopy. Notably, AWESAM astrocytes also exhibit spontaneous Ca2+ signaling similar to astrocytes in vivo.
Introduction
Astrocytes influence brain function through trophic support, blood flow, synaptic signaling and plasticity, and intercellular communication – all of which are mechanisms that center around the thin astrocytic processes. The AWESAM protocol described here allows the study of these processes without interference from neurons and other glia, which is useful e.g., because the expression of many proteins and even Ca2+ signaling overlap across different brain cell types. Further, this method overcomes limitations of previously available techniques. In particular, our protocol provides in vivo-like morphology (stellate astrocytes with thin processes) and gene expression in a quick, easy, and cheap manner.
Cell morphology, gene expression, and many other regulatory processes are directly influenced by the environment. In a culture dish, this refers to factors released by surrounding cells, but also the medium in which the cells are grown. For astrocytes, HBEGF was previously reported to induce a stellate morphology1 but was also found to de-differentiate astrocytes2. However, lower concentrations of HBEGF were later used for generating more in vivo-like astrocytes as identified via RNA sequencing in two different protocols3,4. Moreover, astrocyte morphology changes with the medium composition, regardless of the protocol4: Neurobasal medium with a low concentration of HBEGF is optimal for growing stellate astrocytes, while other medium compositions (e.g., serum-containing DMEM) produce polygonal cells (even in astrocytes freshly isolated by immunopanning).
The AWESAM protocol overcomes disadvantages of previous techniques for growing astrocyte monocultures4. Previous techniques for growing astrocyte monocultures have the following disadvantages: polygonal morphology, which is uncharacteristic of stellate astrocytes in vivo (MD method)5; the length and cost of protocol (preparing astrocytes from induced pluripotent stem cells takes three months and requires many reagents, including expensive growth factors)6; the low amounts of material (immunopanning method)3; and the de-differentiation of astrocytes and requirement of a 3D matrix (Puschmann et al.)1,2. In contrast, the AWESAM protocol provides: in vivo-like morphology (stellate astrocytes with thin processes); a quick, easy, and cheap method; large quantities of material; and the most in vivo-like gene expression compared with immunopanned and MD astrocytes. Additionally, 2D culture allows for the study of Ca2+ signaling and vesicle recycling in thin processes, and in events close to the membrane (e.g., TIRF microscopy is not possible in 3D cultures).
The MD method has been extensively used since its publication in 19805, offering a simple and fast technique for polygonal astrocyte monocultures. In brief, the MD method entails growing mixed brain cells in fetal calf serum (FCS)-containing DMEM, followed by shaking steps that enrich for astrocytes (as all other brain cell types detach from the dish) and further culture in the same medium. DMEM supplemented with FCS is used for many other cell types, ranging from fibroblasts and adipocytes to different cancer cell lines, all of which share the polygonal morphology exhibited by MD astrocytes in culture. Until the early 2000s7, little thought had been given to media tailored to astrocytes, specifically to favor their typical stellate morphology found in vivo. One protocol published in 2011 does achieve such stellate morphology: Called the immunopanning method, it employs serum-free medium optimized for culturing astrocytes3. Using this method, freshly isolated brain cells are exposed to a series of dishes coated with antibodies that target cell type-specific cell surface proteins, to enrich for astrocytes. Despite the more in vivo-like morphology and expression profile of immunopanned astrocytes, the majority of in vitro studies still rely on the MD astrocyte method. The MD method is simple and fast, while the immunopanning method comprises more complex and time-consuming steps on the first day of culture (such as longer enzyme digestion periods, careful layering of solutions of different density, immunopanning itself, and several centrifugation steps – all before plating). However, by using more specialized medium, the AWESAM method offers both the speed of the MD method and the in vivo-like morphology of the immunopanning protocol.
Overall, the AWESAM protocol is useful for studying more in vivo-like astrocytes in 2D monocultures isolated from neurons and other glia (as previously characterized4 by immunostainings, immunoblots, and RNA sequencing). It allows for the study of thin astrocytic processes, and provides great accessibility for visualizing spontaneous Ca2+ signaling and events close to the membrane (e.g., imaged by TIRF microscopy).
Protocol
All animal experiments described here were performed in accordance with the guidelines for German animal welfare.
NOTE: The timeline and main steps of the protocol are illustrated in Figure 1.
1. Preparations
- Prepare the following solutions.
- Prepare DMEM+ for culturing polygonal MD astrocytes: DMEM with 10% FCS, 100 U penicillin and 100 µg/mL streptomycin.
- Prepare NB+H for culturing stellate astrocytes: neurobasal medium with 5 ng/mL HBEGF, 1x B-27 supplement, 1x glutamine supplement, 100 U penicillin and 100 µg/mL streptomycin.
NOTE: Aliquots of higher concentrations of HBEGF can be prepared and stored at -20 °C to be added to the medium whenever needed. - Prepare the dissection medium (10 mM HEPES in HBSS).
- Prepare aliquots of 6-7 mL of 0.05% trypsin-EDTA and 6-7 mL 0.25% trypsin-EDTA in 15 mL tubes. These can be stored at -20 °C and thawed in a 37 °C water bath.
- The day before starting the protocol, coat 10 cm tissue culture-treated dishes with 5 mL of 0.04% polyethylenimine (PEI) each.
- The next day, remove the PEI and wash the dishes with deionized, distilled water twice. Add 12 mL of DMEM+ to each plate and pre-equilibrate in the incubator at 37 °C and 5% CO2 for at least 30 min before plating cells.
- Prepare the dissection area: In a laminar flow dissection hood, place two 10 cm Petri dishes filled with dissection medium on ice. For each brain area to be isolated (e.g., cortex, hippocampus), prepare a 15- tube filled with 14 mL dissection medium and keep on ice. Spray dissection tools (fine scissors, Dumont #3 and #5 forceps) with 70% ethanol and place next to dissection microscope in the hood.
2. Rat or Mouse Brain Dissection
NOTE: Both mice and rats can be used. Animals older than P8 may also be used (we tested up to P12), but removing the meninges will become increasingly difficult. Further, the cell yield and health will diminish, since brain cells from older animals will have formed intricate processes and connections that may break during tissue dissociation, leading to increased cell death. Cortical preparations are described here, but other brain regions can also be used.
- Place 0.05% trypsin-EDTA in a 37 °C water bath.
- Euthanize animals (pregnant rats for E19 pups, or P0-P8 animals) by slowly raising CO2 concentration, and isolate the brains. The following steps require sterile technique.
- If working with embryonic tissue, first remove the embryos from the uterus and open the embryonic sacs, then immediately cut off the heads with scissors. When all heads are cut off, transfer them into pre-cooled dissection medium.
- Once the heads are submerged in the medium, open the skin and skull (both are very soft in young animals) with forceps and pinch off the cerebellum. Carefully lever the rest of the brain up and out of the skull.
NOTE: Two to four E19 embryos yield enough tissue for at least eight 10 cm dishes of cortical astrocytes plated at 500,000 cells per dish on the day of dissection.
- Once the heads are submerged in the medium, open the skin and skull (both are very soft in young animals) with forceps and pinch off the cerebellum. Carefully lever the rest of the brain up and out of the skull.
- For newborn or older pups, cut off the heads with scissors, and isolate the brains by medially cutting the skin in a straight line from the back of the head to the tip of the nose. Pull the skin aside and carefully cut the skull left and right, then lift the skull bone up. Cut off the cerebellum, and lever the rest of the brain up and out of the skull.
NOTE: Two to three P0-P8 pups yield enough tissue for at least eight 10 cm dishes of cortical astrocytes plated at 500,000 cells per dish on the day of dissection. - Separate the cortex from the underlying midbrain structure. Place the forceps within the longitudinal fissure between the hemispheres, then move the forceps in between the cortex and midbrain structures of one hemisphere (perpendicular to the longitudinal fissure) to pinch each hemisphere free.
- Using the forceps, remove all meninges from each hemisphere (and its hippocampus), then separate the hippocampi. If hippocampal astrocytes are required, move each hippocampus into a 15 mL tube filled with 14 mL dissection medium on ice.
- Remove further areas from each hemisphere's cortex if specific regions are required (e.g., frontal cortex). Cut any cortical tissue to be used for generating astrocytes into pieces no larger than 1 mm3. Collect all pieces of cortical tissue in a separate 15-mL tube filled with dissection medium on ice.
3. Tissue Digestion and Dissociation
- Once all tissue pieces are collected, wait for them to settle to the bottom of the tube, then aspirate as much medium as possible.
- Add 2-3 mL of 0.05% trypsin-EDTA using a serological pipette, and incubate the tubes in a 37 °C water bath for 20 min.
NOTE: Release all trypsin-EDTA from the pipette swiftly to mix the tissue pieces and allow to settle again. - Carefully aspirate the trypsin-EDTA, leaving tissue pieces in as little liquid as possible, at the bottom of the tube.
- Add 5 mL of pre-cooled dissection medium to wash off the remaining trypsin-EDTA. Pipette to the side of the upper part of the tube to achieve mixing of the tissue pieces.
- Aspirate the dissection medium, and leave the tissue pieces in as little liquid as possible. Repeat this washing step with pre-cooled dissection medium twice more.
- After the final washing step, add 1 mL of DMEM+ (warmed to room temperature) using a 1,000 µL pipette tip, and triturate the tissue by pipetting up and down without introducing bubbles.
NOTE: It is important to avoid producing bubbles, as this may change the pH of the solution, to which astrocytes are quite sensitive. - Place a 100 µm cell strainer in the opening of a 50-mL tube and pre-wet the filter with 4.5 mL of pre-cooled DMEM+.
- Pipette the 1 mL of dissociated tissue suspension onto the cell strainer and add another 4.5 mL pre-cooled DMEM+ to wash the cells through the cell strainer.
4. Cell Plating for Astrocyte Enrichment by Passaging
- Count the cells, e.g., by diluting 10 µL of the cell suspension with 10 µL trypan blue and pipetting the mix onto a hemocytometer.
- Plate 500,000 cells in 12 mL DMEM+ per 10-cm dish.
NOTE: DMEM+ is helpful for astrocytic growth, more so than for neurons or microglia. During shaking, the physical forces acting on glass coverslips within wells are much stronger than those acting on the 10-cm dishes (without coverslips). If cells are shaken on 24-well plates, they become unhealthy or die. Only plate astrocytes on 24-well plates after shaking. - Keep cell cultures in the incubator at 37 °C and 5% CO2 for seven days.
- On day 6, the day before shaking and passaging cells, prepare the PEI-coated culture plates as described in step 5.
5. Passaging Cells
- The day before passaging, coat cell culture plates with 0.04% PEI.
- For immunocytochemistry, viral transduction, or plasmid transfection, use 24-well or 6-well plates (each containing a sterilized 12 mm or 25 mm glass coverslip, respectively).
- Sterilize the glass coverslips by incubation in 70% ethanol for at least 1 h, then wash three times in deionized, distilled water before placing the coverslips in the wells.
- Add at least 70 µL 0.04% PEI per 12-mm coverslip in 24-well plates, and 600 µL 0.04% PEI per 25 mm coverslip in 6-well plates.
- For Western blot or RNA-Seq analysis, the 10 cm dishes already containing the cells after the dissection can continue to be used. Dishes only need to be shaken, then the cells are washed with 1x PBS and treated with NB+H (do not transfer to new dishes).
- On day in vitro (DIV) 7 after the dissection, place the 10-cm dishes with cells in DMEM+ on a laboratory shaker in the incubator and shake the dishes at 110 rpm for 6 h.
- Wash off the PEI from coated coverslips with deionized, distilled water twice. Add NB+H medium to the plates (500 µL per well for 24-well plates and 2 mL per well for 6-well plates). Pre-equilibrate the plates in the incubator at 37 °C and 5% CO2 while the 10 cm dishes are shaking, or for at least 30 min.
- 20-30 min before the end of the 6 h shaking, pre-warm 1x PBS, DMEM+, NB+H, and 0.25% trypsin-EDTA to 37 °C in the water bath.
- After 6 h of shaking, take the culture dishes off the shaker and immediately replace the medium (that contains shaken-off neurons, oligodendrocytes, and microglia) with 10 mL pre-warmed 1x PBS per dish.
NOTE: It is important to immediately aspirate the old medium containing detached cells to avoid the non-astrocytic cells re-attaching. - Remove the PBS and add 3 mL pre-warmed trypsin-EDTA per dish. Incubate each dish for 4 min in the incubator at 37 °C and 5% CO2.
NOTE: If preparing samples for Western blot or RNA-Seq, do not add trypsin-EDTA. Instead, replace the PBS with 12 mL pre-warmed NB+H, after which cultures can be placed back in the incubator. - Add 5 mL pre-warmed DMEM+ to the 3 mL trypsin-EDTA in each dish, pipette the cells off the dish using a 5 mL serological pipette, and collect the cell suspension in a 50 mL tube.
NOTE: Pipette directly from the bottom of the dish – the floor of the dish should become clearer as the cells detach. Only use serum-containing medium (in place of DMEM+), as serum inactivates trypsin. - Centrifuge the cell suspension at 3,220 x g at 20 °C for 4 min.
- Remove the supernatant and resuspend the cell pellet in 1 mL pre-warmed NB+H.
6. Cell Plating for Final Astrocyte Cultures
- Count the cells.
- Plate 20,000 cells per well in 6-well plates (in 2 mL NB+H medium per well) or 5,000 cells per well in 24-well plates (in 500 µL NB+H medium per well).
- Keep the cell cultures in the incubator at 37 °C and 5% CO2 until further treatment or analysis.
- Exchange half the medium once a week.
NOTE: In low-nutrient environments, astrocytes stop proliferating but any remaining microglia will proliferate (although in our experience, no microglia appeared in AWESAM cultures). Exchanging half the medium once a week prevents enrichment of any potentially remaining microglia8, and the monocultures remain healthy for at least three weeks.
Representative Results
Astrocytes that are co-cultured with neurons and grown in NB+ medium appear stellate after 2 weeks of culture (Figure 2). In addition to NB+ medium, the co-cultured astrocytes are also exposed to unknown neuron-derived factors that likely contribute to their survival and morphology. In contrast, MD astrocytes grown in DMEM+ as monocultures (after shaking off other cell types before passage) appear polygonal after 2 weeks. AWESAM astrocytes grown in NB+H after shaking, appear stellate with many fine processes after 2 weeks of culture.
We previously showed that the RNA expression profile of AWESAM astrocytes is closer to that of astrocytes in vivo when compared to MD and immunopanned astrocytes4. In terms of astrocyte-specific protein expression, cortical astrocytes express high levels of ALDH1L1 and low levels of GFAP in vivo12. This also applies to AWESAM astrocytes, and the immunostaining of ALDH1L1, as a cytoplasmic enzyme, reveals finer detail than the cytoskeletal protein GFAP (Figure 2B).
Aside from the RNA and protein expression, astrocytes in vivo show spontaneous Ca2+ signaling (i.e., without external stimulation), but polygonal MD astrocytes in vitro often require stimulation to elicit Ca2+ signaling, e.g., by adding ATP or glutamate. AWESAM astrocytes exhibit spontaneous Ca2+ signaling in vivo, in both cell bodies and fine processes, which can be visualized by virally transducing cells with the membrane-targeted Lck-GCaMP3 (Figure 3). Spontaneous waves of Ca2+ throughout adjoining astrocytes also occur (Figure 3A). Astrocytic Ca2+ signaling (Figure 3B) in fine processes and microdomains can be analyzed using freeware specific to astrocytes13.
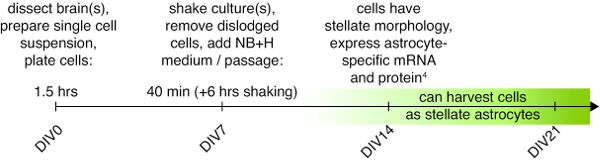
Figure 1: Protocol timeline. A timeline showing the main steps of the AWESAM protocol, including the time it takes to perform these steps on DIV0 (day of dissection), DIV7 (day of passage), and what to expect at later time points. The green box illustrates when cells can be harvested as stellate astrocytes, where lighter versus darker shades represent less versus more complex stellate morphology and maturity, respectively. Please click here to view a larger version of this figure.
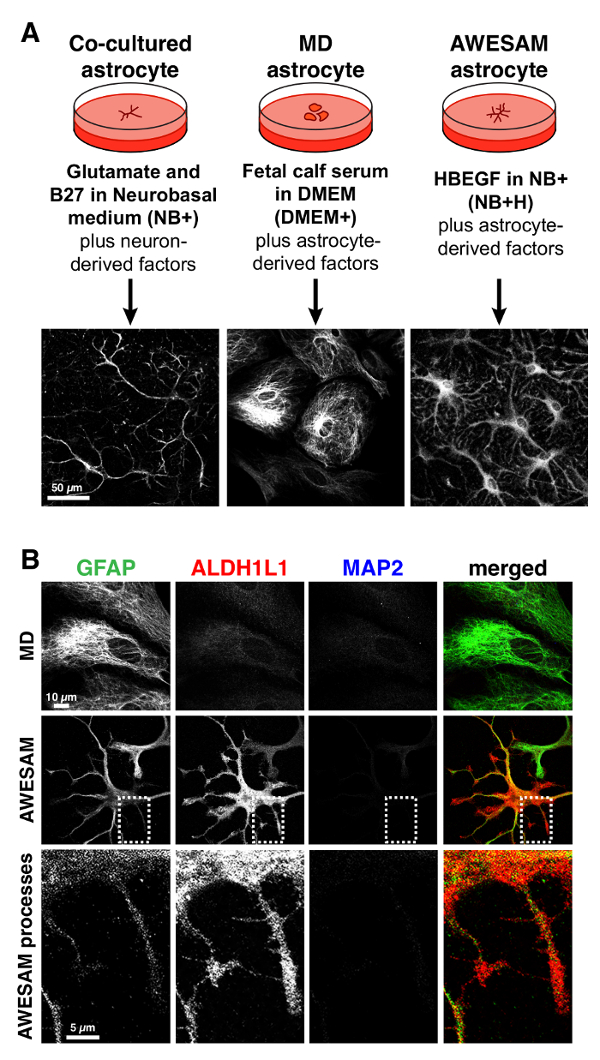
Figure 2: Medium determines morphology. (A) Schematic showing how astrocyte morphology changes when co-cultured with neurons (as previously described4) vs. grown as monocultures, and depending on the medium. Astrocyte-specific staining with GFAP outlines the stellate morphology in co-cultured and AWESAM astrocytes, and the polygonal morphology in MD astrocytes. (B) GFAP labels MD astrocytes more strongly than ALDH1L1, and ALDH1L1 (but not GFAP) outlines the fine processes of AWESAM astrocytes. Neither astrocyte monoculture exhibits the neuronal marker MAP2. Please click here to view a larger version of this figure.
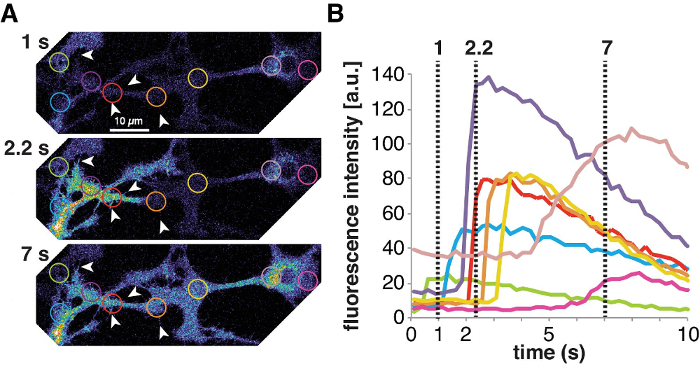
Figure 3: Spontaneous Ca2+ signaling in astrocytes. (A) GCaMP-transduced AWESAM astrocytes exhibit spontaneous Ca2+ events throughout astrocyte networks, including in the thin processes (arrowheads). A Ca2+ wave travels through several astrocytes from left to right in images from distinct time points, where light color depicts areas of high Ca2+ concentrations, and black areas represent extracellular regions. Images were acquired using live cell confocal microscopy. (B) Ca2+ concentrations change in the color-coded regions in (A) over time, shown as GCaMP fluorescence intensity traces that illustrate how a Ca2+ signaling wave spreads across several astrocytes. Please click here to view a larger version of this figure.
Discussion
Four steps within the protocol are critical: 1) preparing the HBEGF concentration to precisely 5 ng/mL, since small increases in the concentration can lead to immature cultures, e.g., 10 ng/mL HBEGF will de-differentiate astrocytes4; 2) avoiding bubbles in the solutions that contain tissue or cells, which can change the pH and impede astrocyte health; 3) pre-equilibrating media to achieve the optimal pH and temperature for growing healthy astrocytes; 4) exchanging only half the medium once a week because any remaining microglia are less likely to proliferate when sufficient nutrients are present and the medium is exchanged regularly8 (although no microglia were found in AWESAM cultures so far). For this last point, it is important to note that astrocyte-derived factors within the culture should not be entirely removed, as they may support astrocytic survival, differentiation, and proliferation.
It is important to use DIV14 or older cultures as the mature astrocyte source (but for transduction or transfection steps, earlier cultures can be used) because astrocytes younger than DIV14 still have neural stem cell potential9. For viral transduction, mixed cultures of neurons and astrocytes transduced on DIV7 (using adeno-associated viruses) efficiently express virally transduced GFP10. For plasmid transfection, DIV10-DIV24 astrocytes are often used and fluorophore-tagged proteins imaged 2-5 days later11. In our experience, both viral transduction and plasmid transfection work best if astrocytes are transduced between DIV9 and DIV14. The transfection and transduction efficiency depend on the construct and method, but we find that transfection efficiency is ~20% in astrocyte monocultures using lipofectamine transfection, and adeno-associated virus (AAV) transduction efficiency is ~90% (see our previous work4 for further details, e.g., construct used). When transducing astrocytes with AAV particles and transfecting by lipofectamine, we allowed at least 5 and 2 days, respectively, for expression of the construct. Before transduction or transfection, the astrocytes should also be allowed to recover for at least one day after passaging.
Moreover, when preparing cultures for comparing RNA/protein expression at different time points, cultures can be plated at different densities to achieve similar amounts of material, e.g., astrocyte monocultures to be harvested at DIV7 and DIV21 can be plated at densities of 1 x 106 and 500,000 cells per 10-cm dish, respectively. For example, the whole protein lysate yield will be around 0.5 mg/mL on DIV14 when harvested after initially plating 500,000 AWESAM astrocytes on a 10-cm dish.
The AWESAM protocol represents a fast, simple, and inexpensive way to culture astrocytes in isolation from neurons but with in vivo-like characteristics of RNA and protein expression, morphology, and Ca2+ signaling4, which thus far is provided by no other protocol. AWESAM astrocytes can be used for analyses that are typical for cell culture studies, including immunocytochemistry, immunoblotting, RNA expression analysis, TIRF microscopy, and Ca2+ imaging4. In particular, recent advances in genetically encoded Ca2+ indicators allow for visualizing fine astrocytic processes in greater detail than classical Ca2+ dyes14. Ca2+ signaling, vesicular uptake, and exocytosis in both thick and thin astrocytic processes is physiologically relevant, as these processes can influence brain function in terms of trophic support, blood flow, synaptic signaling and plasticity, and intercellular communication. The AWESAM protocol allows for the study of the fine astrocytic processes relevant for brain physiology15 and disease16,17, without neuronal interference and with the great accessibility that in vitro culture supplies.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
We acknowledge funding from the European Research Council (FP7/260916), the Alexander von Humboldt-Stiftung (Sofja Kovalevskaja), and the Deutsche Forschungsgemeinschaft (DE1951 and SFB889).
Materials
| 0.05% trypsin-EDTA | Gibco | 25300054 | warm in 37 ºC waterbath before use |
| 0.25% trypsin-EDTA | Gibco | 25200-056 | warm in 37 ºC waterbath before use |
| B-27 supplement | Gibco | 17504-044 | 50X stock |
| Glutamax | Gibco | 35050-061 | 100X stock |
| Penicillin / streptomycin | Gibco | 15070-063 | 50 stock; penicillin: 5000 U/ml; streptomycin: 5000 µg/ml |
| Neurobasal medium | Gibco | 21103049 | without L-glutamine |
| DMEM | Gibco | 41966029 | |
| HBEGF | Sigma | 4643 | |
| HEPES | Gibco | 15630080 | |
| HBSS | Gibco | 14170112 | |
| FCS | Gibco | 10437028 | |
| PBS | Gibco | 10010049 | 1X |
| 100 μm nylon cell strainer | BD | 352360 | |
| Trypan Blue | Sigma | T8154 | |
| Cell culture incubator | Thermo Fisher Scientific | Hera Cell 240i cell culture incubator | |
| Laboratory shaker | Heidolph | Rotamax 120 | use inside cell culture incubator |
| Centrifuge | Eppendorf | 5810 R |
Riferimenti
- Puschmann, T. B., et al. Bioactive 3D cell culture system minimizes cellular stress and maintains the in vivo-like morphological complexity of astroglial cells. Glia. 61 (3), 432-440 (2013).
- Puschmann, T. B., et al. HB-EGF affects astrocyte morphology, proliferation, differentiation, and the expression of intermediate filament proteins. J Neurochem. 128 (6), 878-889 (2014).
- Foo, L. C., et al. Development of a method for the purification and culture of rodent astrocytes. Neuron. 71 (5), 799-811 (2011).
- Wolfes, A. C., et al. A novel method for culturing stellate astrocytes reveals spatially distinct Ca2+ signaling and vesicle recycling in astrocytic processes. J Gen Physiol. 149 (4), (2016).
- McCarthy, K. D., de Vellis, J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 85 (3), 890-902 (1980).
- Krencik, R., Zhang, S. -. C. Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nat Protoc. 6 (11), 1710-1717 (2011).
- Morita, M., et al. Dual regulation of calcium oscillation in astrocytes by growth factors and pro-inflammatory cytokines via the mitogen-activated protein kinase cascade. J Neurosci. 23 (34), 10944-10952 (2003).
- Saura, J. Microglial cells in astroglial cultures: a cautionary note. J Neuroinflammation. 4, 26 (2007).
- Raponi, E., et al. S100B expression defines a state in which GFAP-expressing cells lose their neural stem cell potential and acquire a more mature developmental stage. Glia. 55 (2), 165-177 (2007).
- Royo, N. C., et al. Specific AAV serotypes stably transduce primary hippocampal and cortical cultures with high efficiency and low toxicity. Brain Res. 1190, 15-22 (2008).
- Malarkey, E. B., Parpura, V. Temporal characteristics of vesicular fusion in astrocytes: examination of synaptobrevin 2-laden vesicles at single vesicle resolution. J Physiol. 589, 4271-4300 (2011).
- Zamanian, J. L., et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2 (18), 6391-6410 (2012).
- Srinivasan, R., et al. Ca(2+) signaling in astrocytes from Ip3r2(-/-) mice in brain slices and during startle responses in vivo. Nat Neurosci. 18 (5), 708-717 (2015).
- Shigetomi, E., et al. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J Gen Physiol. 141 (5), 633-647 (2013).
- Srinivasan, R., et al. New Transgenic Mouse Lines for Selectively Targeting Astrocytes and Studying Calcium Signals in Astrocyte Processes In Situ and In Vivo. Neuron. 92 (6), 1181-1195 (2016).
- Otsu, Y., et al. Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci. 18 (2), 210-218 (2015).
- Agarwal, A., et al. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron. 93 (3), 587-605 (2017).
