A Melanoma Patient-Derived Xenograft Model
Summary
Patient-derived xenograft (PDX) models more robustly recapitulate melanoma molecular and biological features and are more predictive of therapy response compared to traditional plastic tissue culture-based assays. Here we describe our standard operating protocol for the establishment of new PDX models and the characterization/experimentation of existing PDX models.
Abstract
Accumulating evidence suggests that molecular and biological properties differ in melanoma cells grown in traditional two-dimensional tissue culture vessels versus in vivo in human patients. This is due to the bottleneck selection of clonal populations of melanoma cells that can robustly grow in vitro in the absence of physiological conditions. Further, responses to therapy in two-dimensional tissue cultures overall do not faithfully reflect responses to therapy in melanoma patients, with the majority of clinical trials failing to show the efficacy of therapeutic combinations shown to be effective in vitro. Although xenografting of melanoma cells into mice provides the physiological in vivo context absent from two-dimensional tissue culture assays, the melanoma cells used for engraftment have already undergone bottleneck selection for cells that could grow under two-dimensional conditions when the cell line was established. The irreversible alterations that occur as a consequence of the bottleneck include changes in growth and invasion properties, as well as the loss of specific subpopulations. Therefore, models that better recapitulate the human condition in vivo may better predict therapeutic strategies that effectively increase the overall survival of patients with metastatic melanoma. The patient-derived xenograft (PDX) technique involves the direct implantation of tumor cells from the human patient to a mouse recipient. In this manner, tumor cells are consistently grown under physiological stresses in vivo and never undergo the two-dimensional bottleneck, which preserves the molecular and biological properties present when the tumor was in the human patient. Notable, PDX models derived from organ sites of metastases (i.e., brain) display similar metastatic capacity, while PDX models derived from therapy naive patients and patients with acquired resistance to therapy (i.e., BRAF/MEK inhibitor therapy) display similar sensitivity to therapy.
Introduction
Preclinical models are critical for all aspects of translational cancer research, including disease characterization, discovery of actionable vulnerabilities unique to cancer versus normal cells, and the development of efficacious therapies that exploit these vulnerabilities to increase the overall survival of patients. In the melanoma field, tens of thousands of cell line models have been heavily utilized for drug screening, with >4,000 contributed by our group alone (WMXXX series). These cell line models were derived from melanoma patients with various forms of cutaneous melanoma (i.e., acral, uveal, and superficial spreading) and diverse genotypes (i.e., BRAFV600-mutant and neuroblastoma RAS viral oncogene homolog [NRASQ61R-mutant]), which span the spectrum of disease present in the clinic1,2.
Unequivocally, the most successful, targeted therapy strategy in the melanoma field has emerged from 1) the genomic characterization of patients’ tumors identifying BRAF mutations in ~50% of melanomas3 and from 2) preclinical investigation leveraging melanoma cell line models4. The BRAF/MEK inhibitor combination was Food and Drug Administration (FDA)-approved in 2014 for the treatment of patients whose melanomas harbor activating BRAFV600E/K mutations and boasts a >75% response rate5. Despite this initial efficacy, resistance rapidly arises in nearly every case due to multifarious intrinsic and acquired resistance mechanisms and intratumoral heterogeneity. Unfortunately, cell line models do not recapitulate representative biological heterogeneity when grown in two-dimensional culture in plastic vessels, which masks their clinically predictive potential when investigators attempt to experimentally determine therapies that might be effective in patients with a specific form or genotype of melanoma6. Understanding how to best model patient intratumoral heterogeneity will allow investigators to better develop therapeutic modalities that can kill therapy-resistant subpopulations that drive failure to current standard-of-care therapies.
Paramount to the limited predictive value of cell line models is how they are initially established. Irreversible alterations occur in the tumor clonal landscape when a single-cell suspension of a patients’ tumor is grown on two-dimensional, plastic tissue culture vessels, including changes in proliferative and invasive potential, the elimination of specific subpopulations, and the alteration of genetic information7. Xenografts into mice of these melanoma cell line models represent the most frequently used in vivo platform for preclinical studies; however, this strategy also suffers from the poor recapitulation of complex tumor heterogeneity observed clinically. To overcome this shortcoming, there has been a growing interest in incorporating more sophisticated preclinical models of melanoma, including the PDX model. PDX models have been utilized for >30 years, with seminal studies in lung cancer patients demonstrating concordance between the patients' response to cytotoxic agents and the response of the PDX model derived from the same patient8. Recently, there has been a drive to utilize PDX models as the tool of choice for preclinical investigations both in the industry and in academic centers. PDX models, because of their superior recapitulation of tumor heterogeneity in human patients, are more clinically relevant to use in therapy optimization efforts than cell line xenografts9. In melanoma, there are immense hurdles that blunt the therapeutic management of advanced disease10. Clinically relevant PDX models have been used to model clinical resistance and identify therapeutic strategies with clinically available agents to treat therapy-resistant tumors11,12. Briefly, the protocol presented here to generate PDX models requires the subcutaneous implantation of fresh tissue from primary or metastatic melanomas (collected by biopsy or surgery) into NOD/scid/IL2-receptor null (NSG) mice. Different variations in methodological approach are used by different groups; however, a fundamental core exists13.
Protocol
The following animal protocols follow the guidelines of The Wistar Institute’s humane ethics committee and animal care guidelines.
1. Melanoma tumor tissue collection
- Collect tumor tissue (termed passage 0) from melanoma patients by one of the following surgery or biopsy methods.
- For surgical excision tissue, maintain a minimum of 1 g of tissue (resect metastases and primary lesions) in transport storage media (RPMI 1640 + 0.1% fungizone + 0.2% gentamicin) at 4 °C or on ice.
- For surgical biopsy tissue, maintain less than 1 g of tissue (often punch biopsies of subcutaneous [s.c.] metastases and lymph node [LN] metastases) in transport storage media at 4 °C or on ice.
- For core biopsy tissue, wash out a cylinder (core) of tissue of approximately 10 mm x 1 mm (often, liver biopsies) into a 15 mL tube containing 5 mL of transport storage media at 4 °C or on ice.
- For fine needle aspirate (FNAs) tissue, keep a very small amount of tissue (less than 1 mm in size) taken directly from the patient in a needle and syringe at 4 °C or on ice.
- Deliver the tissue in transport storage media at 4 °C or on ice on the same day or with overnight shipping after surgical excision or biopsy. Process the tissue within 1-2 h of delivery.
2. Tumor tissue processing for mouse implantation
- Surgical excision or surgical biopsy tissue processing
- Transfer the tissue to a sterile Petri dish and separate the tumor tissue from surrounding normal tissue as much as possible.
- Remove necrotic tissue (usually identified as pale-whitish tissue located centrally within the tumor) from the remaining tumor as much as possible.
- Use a scalpel to subdivide an initial tumor chunk into approximately equal pieces (~3 mm x 3 mm) for surgical mouse implantation (Figure 2).
- Optionally, if enough tumor tissue is available, snap-freeze the tissue for downstream assays (RNA sequencing [RNASeq], whole exome sequencing [WES], etc.).
- Make a tumor slurry by mincing the tumor tissue using a cross blade technique with two scalpel blades. Mince the tumor chunks as finely as possible to form a slurry, which is now ready for surgical mouse implantation.
- Alternatively, if the tumor tissue is too hard for mechanical dissociation, use a digestion dissociation procedure to form gel-like slurry and a single-cell suspension for implantation and/or injection.
- Mince the tumor chunks as finely as possible to form slurry.
- Put the slurry in a 50 mL tube with cold Hank’s balanced salt solution (HBSS)-/- (without Ca++ and Mg++); then, centrifuge and pellet at 220 x g for 4 min at 4 °C.
- Resuspend the slurry in 10 mL of warmed fresh digest media (200 U/mL collagenase IV + 5 mM CaCl2 + 50 U/mL DNase in HBSS-/-) per 1 g of tumor tissue.
- Place the tube in a 37 °C water bath for 20 min and mix vigorously every 5 min with a disposable pipette.
- Wash with up to 50 mL of HBSS-/-; then, centrifuge at 220 x g for 4 min at 4 °C.
- Add 5 mL of prewarmed TEG (0.025% trypsin + 40 µg/mL ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid [EGTA] + 10 µg/mL polyvinyl alcohol [PVA]) per 1 g of tumor tissue, gently resuspend/shake, and place the tube at 37 °C for 2 min without mixing.
- Add at least 1 equal volume of cold staining media (1% bovine serum albumin [BSA] + 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES] + 1x penicillin-streptomycin in L15 media) to quench the trypsin and centrifuge at 220 x g for 4 min at 4 °C.
- Resuspend the sample in 10 mL of staining media per 1 g of tumor tissue and filter it through a 40 µm cell strainer to get a single-cell suspension for mouse injection (Figure 2).
- Slurry remaining on top of the cell strainer can also be collected for surgical mouse implantation.
- Core biopsy tissue processing
- Pour the core cylinder tissue in tissue-transport tube into a 5 cm Petri dish.
- Remove excess liquid, scrape tissue to the edge of the Petri dish, finely mince the tumor tissue, add ~100–150 µL of HBSS-/- on top of the tumor tissue, and quickly draw the HBSS/tumor tissue suspension into the 1 mL syringe.
- Attach a 23 G needle to the 1 mL syringe, pass the HBSS/tumor tissue suspension through the needle into a 1.5 mL spin tube.
- Redraw the tumor suspension into the syringe with the needle still on until it can pass smoothly through the needle.
- Finally draw the tumor suspension back into the syringe and detach the 23 G needle.
- Attach a 27 G needle and draw an equal volume (~100–150 µL) of artificial extracellular matrix (see Table of Materials) into the syringe.
- Add an equal volume of artificial extracellular matrix slowly into the 1.5 mL spin tube with the tumor/HBSS-/- suspension, carefully avoiding the formation of bubbles.
- Draw one last time back into the syringe and the core biopsy tumor tissue is now ready for mouse injection.
- FNA tissue processing
- Place the syringe containing the FNA samples (trapped in the needle) on ice.
- Separate the needle (containing FNA tissue) from the syringe and remove the plunger from the syringe, add ~150–200 µL of HBSS-/- to the top of the syringe.
- Re-insert the plunger into the syringe and the needle (containing FNA tissue), and push out the HBSS-/- through the needle into a 1.5 mL spin tube. This step removes the FNA tumor from the needle.
- Repeat step 2.3.3 with the HBSS-/-/FNA suspension twice using the same syringe to maximize the retrieval of tumor tissue from the needle.
- Add an equal volume (~100–150 µL) of artificial extracellular matrix to the 1.5 mL spin tube containing the HBSS/FNA suspension.
- Adequately mix by slowly drawing up the artificial extracellular matrix/HBSS-/-/FNA suspension back into the 23 G needle and repeating twice.
- Draw the entire artificial extracellular matrix/HBSS-/-/FNA volume back into the syringe, replace the 23 G needle with a 27 G needle, and the FNA sample is now ready for mouse injection.
3. Tumor implantation and injection in mice
- Implantation of surgical excision or surgical biopsy tissue
NOTE: Ensure all surgical instruments are sterile by autoclaving or the use of pre-sterilized disposable instruments.- Shave hair from the lower back of NSG 6-8 week male or female mice leaving an approximately 1.5 cm x 3 cm area with no hair. Anesthetize mice using isoflurane, and confirm by gently squeezing the foot as a test of responsiveness. Use vet ointment on their eyes to prevent dryness.
- Place individual mice on a heat pad in the nose cone of the anesthesia machine, scrub the shaved area with chlorhexidine. Then douse with 70% ethanol and allow to evaporate.
- Prepare chunks or divide tumor slurry in a Petri dish into individual mounds for surgical implantation (i.e., into three equal mounds if to be implanted into 3 mice).
- Using the scalpel blade, make an incision of approximately 5 mm long on the center of the back of the mouse, take one pair of forceps and lift up skin on the side of the incision opposite of operator.
- Take the scissors into the other hand and separate the skin from the muscle layer by gently cutting the fascial membrane with small scissor cuts, thereby creating a "pocket" for the tumor tissue.
- Pick up one tumor chunk or one individual mound of tumor slurry tissue with the scalpel blade and gently place tissue into the created pocket.
- Administer 100 µL of artificial extracellular matrix on the tumor tissue mound in the pocket.
- Using two pairs of forceps, pull up the incision on both ends so that the wound edges come close together, and close the wound by applying one or two wound clips.
- Subcutaneous inject 1-5 mg/kg meloxicam as an analgesic in mice after surgery.
- Take the mouse out of the nose cone and place it back into its original cage, observe the mouse while waking up. Do not return to a cage until fully recovered.
- Remove wound clips after approximately 7 days. If healing is not complete after 7 days, leave the wound clip in for an additional one or two days.
NOTE: If using a single cell suspension from surgical excision or surgical biopsy tissue processing, it will mix with artificial extracellular matrix (at 1:1 ratio) for mice injection.
- Injection of FNA or core biopsy tissue
- Place an NSG mouse on a steel grid rack, hold the mouse firmly by the tail, and gently pull the mouse back. It will grasp the grid with its front legs firmly. Alternatively, restrain a mouse in the non-dominant hand and let the flank area visible.
- Disinfect the skin of the flank with alcohol prep swabs, slowly and steadily inject the contents of the syringe under the skin of the mouse.
- Pull out the needle and place the mouse back into its cage.
4. Monitor tumor growth
- Monitor mice once weekly to check for palpable tumors.
- Once tumors are at a measurable size (approximately 50 mm3), use a caliper to record tumor dimensions. Use the following formula to calculate tumor volumes: (width x width x length) / 2.
- Harvest tumor once the tumor volume reaches around 1.5 cm3 (approximately 4–10 weeks). The tumor is now called mouse passage 1 (MP1).
5. Harvest tumor for banking tissue, reimplantation, and experiment/characterization
- Euthanize mouse in a CO2 chamber, check vital signs to confirm death, and then submerge the mouse in a Virkon solution to sterilize the skin for 30 s in the bio-cabinet.
- Use curved scissors and surgical forceps to lift skin adjacent to the tumor and make a horizontal cut.
- Use a blunt separation technique to mobilize the skin on both sides of the tumor and over the tumor, exposing the tumor.
- Use scissors or a scalpel blade to separate the tumor from the fascia.
- Resect the tumor and transfer the tumor to a sterile Petri dish, cut the tumor into small pieces and remove necrotic tissue from the tumor.
- Bank tumor tissue for future implantation.
- Take 2-3 small tumor pieces smaller than ~10 mm x 10 mm and mince them into pieces smaller than ~1 mm x 1 mm.
- Transfer all minced tissue to a 2 mL cryogenic vial, add 1 mL of freezing media (10% DMSO + 90% FBS).
- Mix well and place cryogenic vials into a pre-cooled isopropanol-based cell-freezing container on dry ice.
- Store container in -80 °C freezer overnight and then transfer cryogenic vials to liquid nitrogen (LN2) storage.
- Snap-freeze tissue for downstream assays (RNASeq, WES, etc.).
- Place tumor tissue pieces (~3 mm x 3 mm) into a cryogenic vial and put the cryogenic vial in LN2 immediately. Store vials in -80 °C freezer.
6. PDX therapy trials
NOTE: It will take two expansion phases to grow enough tumor tissue to generate the necessary number of PDX bearing mice for the therapy trial.
- Pick up a cryovial containing banked PDX tissue from LN2, and place the cryovial in a 37 °C water bath until the freezing media containing the tumor tissue is just starting to melt.
- Empty the contents of the cryovial into pre-warmed HBSS-/- in a 50 mL tube, and wash tissue.
- Pellet tissue by centrifugation for 5 min at 1,200 rpm.
- Remove HBSS-/- from the tumor sample by vacuum aspiration with a Pasteur pipet.
- Slide the PDX tissue from the 50 mL tube into a 5 cm or 10 cm Petri dish and place on wet ice.
- Follow the above implantation protocol to implant banked tissue from a cryogenic vial into 5 NSG mice for the first round of PDX tumor expansion.
- Once tumors reach ~600–800 mm3, harvest 1-2 tumors to get a single cell suspension with the above protocol for tumor tissue processing: mechanical dissociation, collagenase digestion dissociation procedure.
- Pellet cells and resuspend in 6 mL of HBSS-/-/artificial extracellular matrix (1:1 ratio), subcutaneous inject ~50 NSG mice, with 100 µL of cell mixture per mouse for the second round of PDX tumor expansion.
- Measure tumor size with calipers biweekly.
- Wait for a tumor size of ~100 mm3 in 3-5 weeks, randomize mice into groups, and start treatment.
- When tumor size reaches the maximum IACUC-approved volume, stop treatment.
- Follow the above harvest tumor protocol to collect snap frozen tumor pieces, tumor pieces for paraffin blocks.
Representative Results
Tumor tissue for melanoma PDX models can come from a variety of different sources and can also be processed per the growth dynamics of individual models and the desired use of the PDX tissue. The priority when establishing a PDX model is to have sufficient material to bank for future use and DNA for characterization (Figure 1).
Once sufficient material is banked, tumor tissue can be expanded in one of three main methods to grow enough tumor to perform a formal therapy study (Figure 2A). Each of the methods described herein will allow for the expansion of tumor from PDXs (Figure 2B). It is our experience that creating a single-cell suspension of tumor cells with the use of enzymatic digestion (collagenase IV) can allow for more rapid tumor growth, and can allow one initial tumor to be expanded into 10 – 20 mice, whereas the tumor chunk and slurry method can only be expanded into 5 – 10 mice (Figure 2C). As has been previously demonstrated in other tumor types, melanoma PDX models often reflect the drug sensitivity the patient displayed when on therapy. Shown here is a representative therapy curve from a melanoma patient with BRAFV600E mutant melanoma who initially responded to a BRAF inhibitor but ultimately relapsed. The PDX derived from this patient also displayed initial sensitivity to BRAF inhibition (Rx1) plus an additional inhibitor (Rx2); however, the tumors ultimately relapsed (Figure 3).
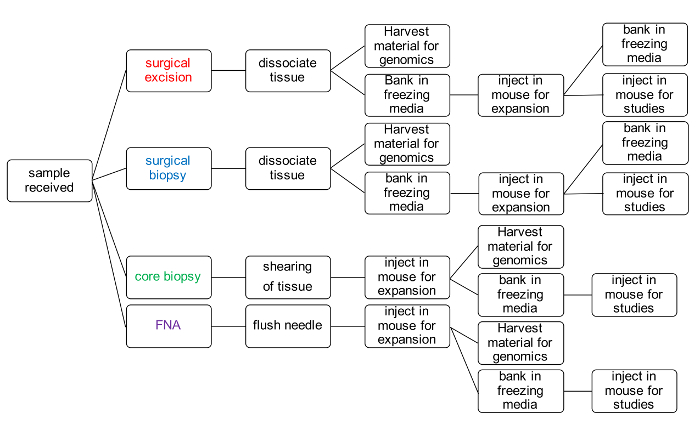
Figure 1: PDX model generation workflow for banking tumor tissue and performing therapy studies. Please click here to view a larger version of this figure.
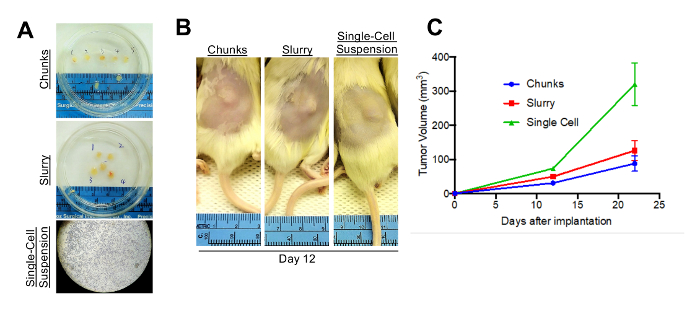
Figure 2: Alternative implantation methods. (A) Tumors can be processed into either chunks, a slurry suspension, or as a single-cell suspension. (B) All three methods will allow for growth of tumors in vivo. Shown here are mice subcutaneously implanted with tumor and imaged 12 days after implantation. (C) Shown are tumor growth curves for the mice injected with one of the three implantation methods. N = 5 per arm; error bars are standard error. Please click here to view a larger version of this figure.
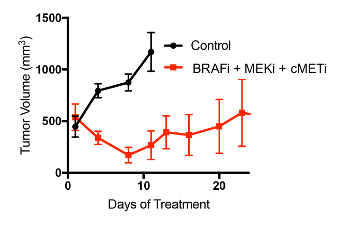
Figure 3: Representative data for a PDX therapy trial. Mice were implanted with PDX tumors and treated with either a vehicle control or a two-inhibitor combination of a BRAF inhibitor and a MEK inhibitor. N=6 per arm. Randomization was used to place mice into study groups. Of note, although ~500 mm3 was used in this example as the starting tumor volume for therapy initiation, the routine volume to begin PDX studies is 100–200 mm3 as PDX tumors are aggressive and their growth is difficult to inhibit once they too big a size (>300 mm3). Please click here to view a larger version of this figure.
Discussion
We have herein described generating PDX models of melanoma with patient tissue derived from primary and metastatic tumors, core biopsies, and FNAs. When directly engrafted into NSG mice, tumors present similar morphologic, genomic, and biologic properties to those observed in the patient. In the case when only a small quantity of tissue is available to investigators, as often occurs with FNAs, the PDX technique allows for the expansion of the tumor tissue for DNA, RNA, and protein characterization, as well as for therapy trials to allow preclinical drug development.
Critical to the success of PDX engraftment is the quality of material investigators begin with. Care must be taken to ensure tumor tissue is appropriately preserved as much as possible in sections 1 and 2. Importantly, the response to therapy of PDX melanoma models better recapitulates the sensitivity of the donor patient, allowing for robust preclinical investigations to develop improved therapeutic strategies to combat therapy resistance and improve the durability of response. Most metastatic melanoma patients do not experience cures with existing standard of care (SOC) therapies5. Our PDX melanoma collection contains more than 500 distinct models, including those derived from patients who relapsed on targeted therapy and immunotherapy1,2. This resource will be critical for the development of therapeutic modalities that overcome resistance to current SOC. Future applications for the PDX model will rely on cost-effective, high throughput approaches that leveraging PDX models in drug screens and CRISPR-Cas9 screens to identify novel effective strategies for different genotypes (i.e., BRAFV600E, NRASQ61R) and subtypes (i.e., uveal, acral) of melanoma14.
One limitation of the PDX technique is the necessity that tumor material is engrafted into mice without an immune system to ensure engraftment success1. Therefore, PDX studies optimizing therapeutic strategies to combat therapy resistance do not address how new therapy strategies may positively or negatively impact the immune system and/or anti-tumor immune responses. Fortunately, advances in the field of mouse humanization with a human immune system have been made and will allow for more ideal PDX studies in mice that better recapitulate the human microenvironment15.
In summary, PDX models allow for preclinical investigations of melanoma cells that better recapitulate the tumor heterogeneity and melanoma aggressiveness observed in the clinic (versus other two-dimensional and standard xenograft approaches). PDX models allow for a deeper understanding of which genes are involved in therapy resistance and provide a more clinically-relevant model from which more effective therapies can be developed to increase the overall survival of patients with metastatic melanoma.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
The authors thank the Wistar Institute Animal Facility, Microscopy Facility, Histotechnology Facility, and Research Supply Center. This study was funded in part by grants from the U54 (CA224070-01), SPORE (CA174523), P01 (CA114046-07), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, and the Melanoma Research Foundation.
Materials
| 1 M Hepes | SIGMA-ALDRICH CORPORATION | Cat # H0887-100ML | |
| 100x PenStrep | Invitrogen | Cat # 15140163 | |
| 1x HBSS-/- (w/o Ca++ or Mg++) | MED | Cat # MT21-023-CV | |
| 2.5% Trypsin | SIGMA-ALDRICH CORPORATION | Cat # T4549-100ML | 10 mL aliquots stored at –20oC |
| BSA | SIGMA-ALDRICH CORPORATION | Cat # A9418-500G | |
| Chlorhexidine | Fisher Scientific | Cat# 50-118-0313 | |
| Collagenase IV (2,000 u/mL) | Worthington | Cat #4189 | make up in HBSS-/- from Collagenase IV powder stock (Worthington #4189, u/mg indicated on bottle and varies with each lot); freeze 1 |
| DMSO | SIGMA-ALDRICH CORPORATION | Cat # C6295-50ML | |
| DNase | SIGMA-ALDRICH CORPORATION | Cat # D4527 | |
| EGTA (ethylene glycol bis(2-aminoethyl ether)-N,N,N’N’-tetraacetic acid) | Merck | Cat # 324626.25 | |
| FBS | INVITROGEN LIFE TECHNOLOGIES | Cat # 16000-044 | |
| Fungizone | INVITROGEN LIFE TECHNOLOGIES | Cat # 15290-018 | |
| Gentamicin | FISHER SCIENTIFIC | Cat # BW17518Z | |
| Isoflurane | HENRY SCHEIN ANIMAL HEALTH | Cat # 050031 | |
| Leibovitz's L15 media | Invitrogen | Cat # 21083027 | |
| Matrigel | Corning | Cat # 354230 | Artificial extracellular matrix |
| Meloxicam | HENRY SCHEIN ANIMAL HEALTHRequisition # ::Henry Schein | Cat # 025115 | 1-5mg/kg, as painkiller |
| NOD/SCID/IL2-receptor null (NSG) Mice | The Wistar Institute, animal facility | breeding | |
| PVA (polyvinyl alcohol) | SIGMA-ALDRICH CORPORATION | Cat # P8136-250G | |
| RPMI 1640 Medium (Mod.) 1X with L-Glutamine | Fisher Scientific | Cat# MT10041CM | |
| Scalpel | Feather | Cat # 2976-22 | |
| Virkon | GALLARD-SCHLESINGER IND | Cat # 222-01-06 | |
| Wound clips | MikRon | Cat #427631 |
Riferimenti
- Garman, B., et al. Genetic and Genomic Characterization of 462 Melanoma Patient-Derived Xenografts, Tumor Biopsies, and Cell Lines. Cell Reports. 21 (7), 1936-1952 (2017).
- Krepler, C., et al. A Comprehensive Patient-Derived Xenograft Collection Representing the Heterogeneity of Melanoma. Cell Reports. 21 (7), 1953-1967 (2017).
- Davies, H., et al. Mutations of the BRAF gene in human cancer. Nature. 417 (6892), 949-954 (2002).
- Paraiso, K. H., et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. British Journal Of Cancer. 102 (12), 1724-1730 (2010).
- Long, G. V., et al. Long-Term Outcomes in Patients With BRAF V600-Mutant Metastatic Melanoma Who Received Dabrafenib Combined With Trametinib. Journal of Clinical Oncology. 36 (7), 667-673 (2018).
- Hidalgo, M., et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discovery. 4 (9), 998-1013 (2014).
- Hausser, H. J., Brenner, R. E. Phenotypic instability of Saos-2 cells in long-term culture. Biochemical and Biophysical Research Communications. 333 (1), 216-222 (2005).
- Fiebig, H. H., et al. Development of three human small cell lung cancer models in nude mice. Recent Results In Cancer Research. 97, 77-86 (1985).
- Izumchenko, E., et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Annals of Oncology. 28 (10), 2595-2605 (2017).
- Shi, H., et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discovery. 4 (1), 80-93 (2014).
- Monsma, D. J., et al. Melanoma patient derived xenografts acquire distinct Vemurafenib resistance mechanisms. American Journal of Cancer Research. 5 (4), 1507-1518 (2015).
- Das Thakur, M., et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 494 (7436), 251-255 (2013).
- Meehan, T. F., et al. PDX-MI: Minimal Information for Patient-Derived Tumor Xenograft Models. Ricerca sul cancro. 77 (21), 62-66 (2017).
- Gao, H., et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nature Medicine. 21 (11), 1318-1325 (2015).
- De La Rochere, P., et al. Humanized Mice for the Study of Immuno-Oncology. Trends in Immunology. 39 (9), 748-763 (2018).
