The methods outlined in this protocol are for the construction of sgRNA and xCas9 expression vectors and then for the optimization screening of sgRNA oligos with relatively higher gene targeting efficiencies. Here we display a representative example of 3 sgRNA targets to sheep DKK2 exon 1. SgRNA and xCas9 expressing vectors can be built by predigesting the vector backbone (Figure 2) followed by ligating it in a series of short double-strand DNA fragments through annealing oligo pairs. The positive colonies could be detected through specific primer pairs guided PCR (Figure 3). An 440 bp DNA fragment from sheep DKK2 exon 1 was subcloned into pSSA-Dual plasmid15 by using double digestion with AscI and SalI, resulting in pSSA-Dual-DKK2. The gene targeting capacities of pX330-xCas9-T1, pX330-xCas9-T2 and pX330-xCas9-T3 were simutanusly detected (Figure 5), and then the last sgRNA vector was identified as the relatively better one, which we pick up for sheep gene editing research at the next step.
This detection method combines a single strand annealing (SSA) mechanism (Figure 1B and Figure 4) with a luciferase report gene in order to monitor DNA cutting efficiency. As illustrated in Figure 4, SSA is a process initiated when a double strand break is made between two repeated sequences oriented in the same direction. Single strand regions are created adjacent to the breaks that extend to the repeated sequences so the complementary strands can anneal to each other. This annealing intermediate can be processed by digesting away the single stranded tails and filling in the gaps. The Dual-Luciferase Reporter gene mainly includes the luciferase genes from the firefly Photinus pyralis and from Renilla reniformis (also known as sea pansy). The activities of firefly and renilla luciferases are measured sequentially from a single sample. When the recognition area that has the termination codon is not cut in its middle, the gene in SSA system is blocked and cannot be translated into a functional protein. When the processing takes place, the SSA system will merge the homologous sequence automatically, and overlapping sequences becomes a single sequence, gene gets recombination repaired and can then be read throughout producing a functional protein.
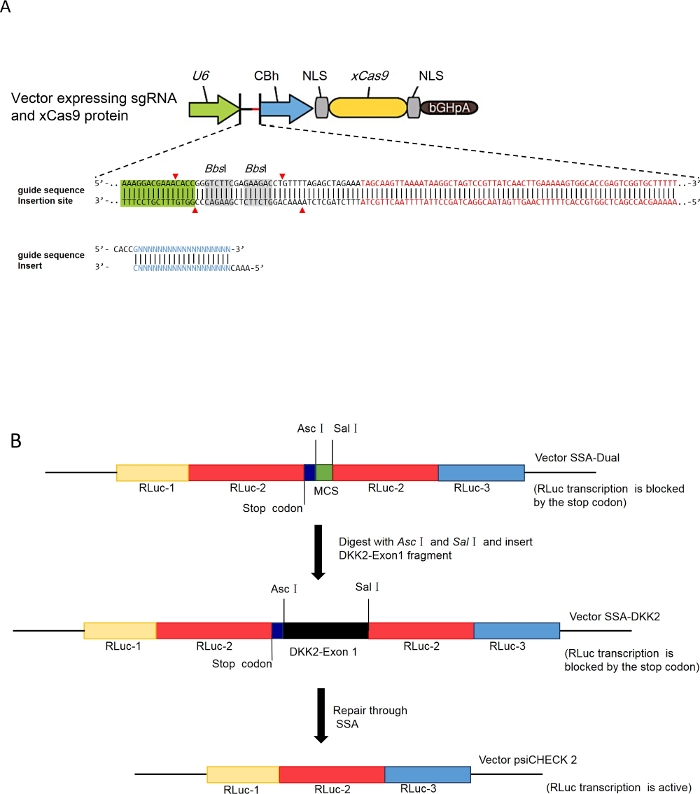
Figure 1: Schematic representation of the different steps of the cloning process.
(A) Schematic of sgRNA vector construction, adapted from the protocols of Feng Zhang Lab. (B) Schematic of dual luciferase reporter vector building (select vector pSSA-DKK2 as an example), DNA fragments Rluc-, Rluc-2 and Rluc-3 represent three parts of full-length coding sequence of Renilla luciferase. MCS represents “multiple cloning site”. Please click here to view a larger version of this figure.
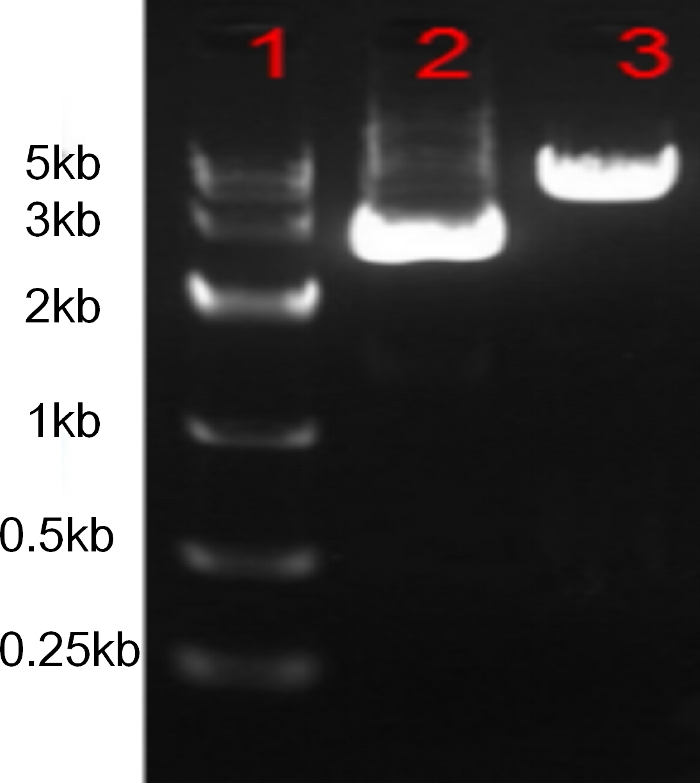
Figure 2: Predigesting the vector backbone pX330-xCas9 with BbsI.
1: DNA Marker, 2: pX330-xCas9 plasmid, 3: pX330-xCas9 plasmid digested with BbsI. Please click here to view a larger version of this figure.
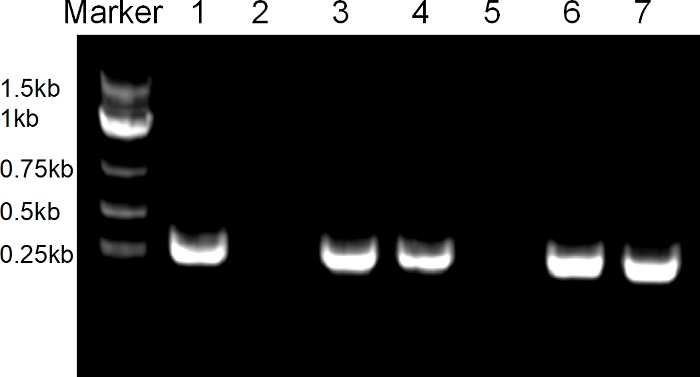
Figure 3: Specific primer pairs guided PCR for DKK2-T1 sgRNA vector detection.
Lanes 1-7 indicate PCR bands for 7 different bacteria colonies separately. Please click here to view a larger version of this figure.
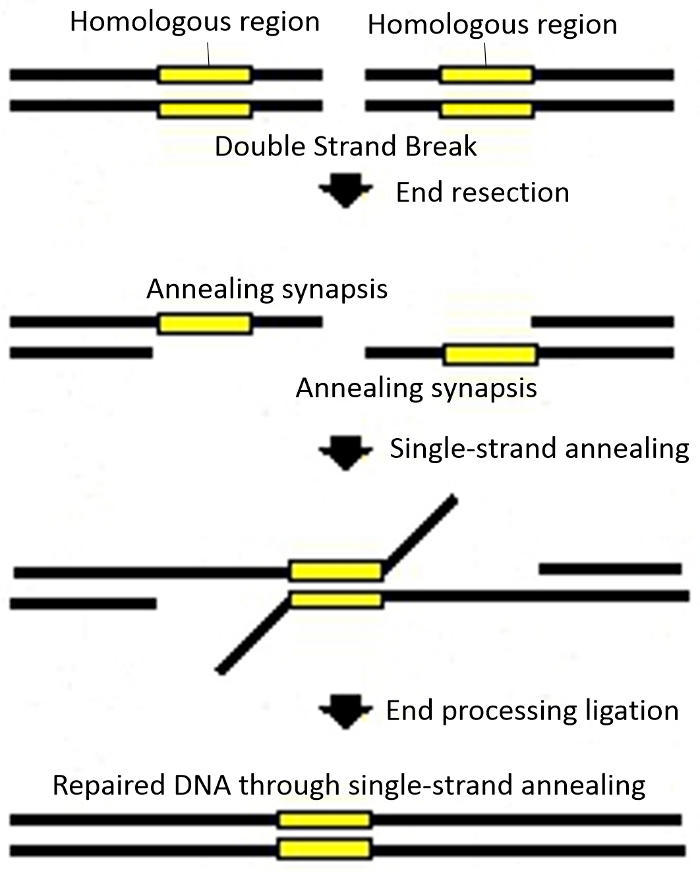
Figure 4: Schematics for single strand annealing (SSA). Please click here to view a larger version of this figure.
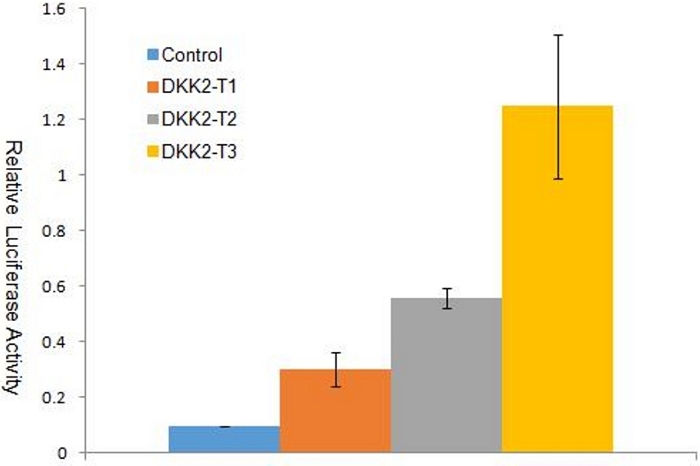
Figure 5: Dual luciferase assay with reporter vector pSSA-DKK2 in cell line PIEC.
The Ranilla luciferase luciferase activities are significantly induced (P<0.01, Student's t test) in PIEC by overexpresing of pX330-xCas9-T1, pX330-xCas9-T2 or pX330-xCas9-T3 with the fold change of 3.15, 5.84 or 13.10, respectively. The error bars indicate standard deviations (SD)(n=3) for each group. Please click here to view a larger version of this figure.
| Name | Genomic DNA Targets (5’-3’) | sgRNA Oligos (5’-3’) | ||
| T1 | TGCCTGCTCCTACTGGCCGCGG | T1-F: CACCGTGCCTGCTCCTACTGGCCGC | ||
| T1-R: AAACGCGGCCAGTAGGAGCAGGCAC | ||||
| T2 | ATCAAGTCCTCTCTGGGCGGGG | T2-F: CACCGATCAAGTCCTCTCTGGGCGG | ||
| T2-R: AAACCCGCCCAGAGAGGACTTGATC | ||||
| T3 | GCCCGCGAGCTGCCGAACTGTG | T3-F: CACCGCCCGCGAGCTGCCGAACTG | ||
| T3-R: AAACCAGTTCGGCAGCTCGCGGGC | ||||
Table 1: sgRNA targets and oligos designed for gene editing of sheep DKK2.
| PCR Component | 25 µL reaction |
| 10 µM Forward Primer (e.g. T1-F) | 1 µL |
| 10 µM Reverse Primer BbsI-R | 1 µL |
| 10x PCR Buffer | 2.5 µL |
| 2.5 mM dNTPs | 1 µL |
| bacterial fluid | 1 µL |
| DNA Taq Polymerase | 2.5 units |
| Nuclease-free water | Up to 25 µL |
Table 2: PCR mixture for positive bacterial colonies detection.
| Reagent | Volume |
| pSSA-Dual vector (synthesized DNA fragment or PCR product) | 1-2 μg |
| CutSmart Buffer | 5 μL |
| AscI | 1 μL |
| SalI-HF | 1 μL |
| Distilled water | up to 50 μL |
| Total volume | 50 μL |
Table 3: Double digestion of pSSA-Dual vector and synthesized DNA fragment (or PCR product).
| Reagent | Volume |
| pSSA-Dual vector(predigested) | 0.5 μg |
| DNA fragments containing the sgRNA targets (predigested) | 0.2 μg |
| T4 DNA Ligase Buffer (10x) | 1 μL |
| T4 DNA Ligase | 1 μL |
| Distilled water | up to 10 μL |
| Total volume | 10 μL |
Table 4: A ligation reaction of DNA fragments containing the sgRNA targets into a predigested pSSA-Dual vector.
| Step 1: reagent preparation | |
| Transfection reagent | 2 μL |
Table 5: Details of cell transfection in a 24 well plate.
