When this protocol is successfully executed, testes will remain fully intact for imaging by confocal microscopy or other fluorescence microscopy methods. As seen in Figure 3A, the cellular organization of the testes is preserved, and the progression of cell differentiation from one end of the testis to the other including spermatogonia, spermatocytes, and haploid spermatids is visible. GFP-tubulin is a useful marker for identifying dividing spermatocytes and for live imaging of the progression of the cells through meiosis. As shown in Figure 3B, spermatocytes about the begin meiosis can be identified by their two prominent microtubule asters. By metaphase the large meiotic spindles are beautifully labeled by GFP-tubulin (Figure 3C).
Drosophila spermatocytes’ large size and their relative slow progression through meiosis make them ideal for analysis of the dynamics and reorganization of cellular components such as cytoplasmic organelles during cell division8,11,21. Figure 4 and Video 1 show live imaging analysis of the dynamic reorganization of the endoplasmic reticulum (ER) with respect to microtubules through the course of meiosis. As described above, during late interphase just prior to the onset of meiosis, two centrosomes located at the cortex of the cell are clearly visible by GFP-tubulin fluorescence. At this stage, ER-targeted RFP (RFP-KDEL) fluorescence reveals that the ER is evenly distributed throughout the cytoplasm. The centrosomes then begin migrating to opposite sides of the nuclear envelope to establish the two spindle poles. During this time, the ER rapidly accumulates around the centrosomal microtubules and is progressively cleared from the rest of the cytoplasm. Nuclear envelope breakdown (NEB) is revealed by the appearance of GFP-tubulin fluorescence within the nuclear region. It should be noted that Drosophila cells divide by semi-open mitosis or meiosis, meaning that nuclear envelope components break down and disperse and large cytoplasmic molecules enter the nuclear region, but an envelope of ER-derived membranes surrounds the spindle and chromatids throughout cell division22,23,24. By the time of NEB and progressing through metaphase, the ER is nearly completely associated with the astral microtubules that surround the two spindle pole centrosomes. These two astral microtubule accumulations of the ER then partition to the two daughter cells during anaphase, ensuring proper ER inheritance by the newly formed cells8,11.
Figure 5 shows the effects of testis rupture on live imaging of spermatocytes. As described in the protocol, great care must be taken not to lower the coverslip too far onto the surface of the testes during the mounting steps, as this will apply pressure to the testes and cause them to burst. As seen in Figure 5 and Video 2, cysts of spermatocytes rapidly flow out of the ruptured testis, and this movement of the cells makes live, time-lapse imaging extremely difficult.
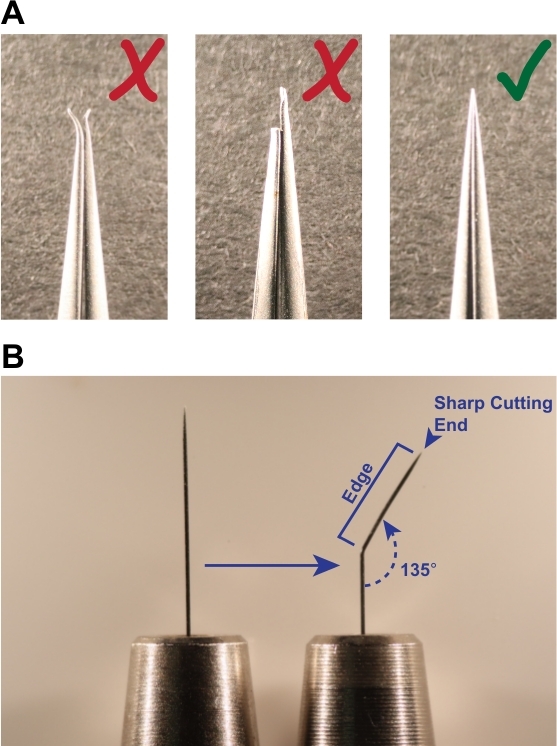
Figure 1: Tools required for successful dissection. (A) Forceps used for dissection must be straight and have sharp, unbroken tips (indicated by green check mark). Do not use forceps with bent or broken tips (red X marks), as they will be ineffective at grasping larvae and teasing apart tissues. (B) To prepare the scalpel tool, bend a black anodized steel insect pin to an internal angle of approximately 135°. The sharp point is used as a knife to cut through tissue, and the straight edge is used for moving tissues. Please click here to view a larger version of this figure.

Figure 2: Identification of larval and pupal testes. (A) Images of male and female larvae. The testes are identifiable in the male as the circular or oval translucent structures in the posterior third of the animal (arrowhead). Note the lack of a similar structure in the female. (B) Image of an early male pupa, with the two translucent testes indicated by arrowheads. (C) Testes with attached fat bodies after dissection. The two testes on the left have excessive fat bodies still attached that need to be trimmed away. The two testes on the right have been properly trimmed of their fat bodies and are ready for imaging. Please click here to view a larger version of this figure.
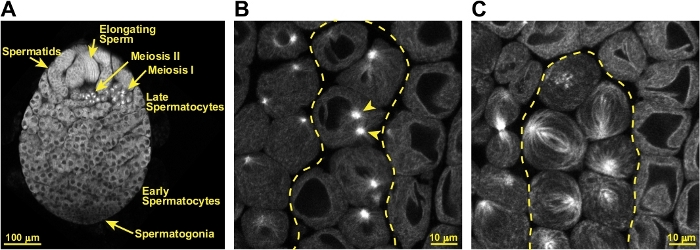
Figure 3: Identification of meiotic spermatocytes in a successfully prepared testis. (A) Confocal image of a successfully prepared live, intact testis expressing GFP-tubulin. The hub of stem cells is located toward the lower tip of the testis, though it is not identifiable in this preparation. Several layers of small spermatogonia are indicated, and these are followed by numerous cysts of spermatocytes. A cyst of spermatocytes in the first meiotic division is identifiable based on the large size of the cells (approximately 20 – 30 µm in diameter) and presence of GFP-tubulin labeled meiotic spindles. Spermatocytes in the second meiotic division similarly have spindles, but these cells are approximately half the size meiosis I cells. Post-meiotic spermatids are also visible, as are elongating spermatozoa. (B) Spermatocytes that will begin meiosis within 30 – 60 min can be identified by their two large, very bright microtubule asters (indicated by arrow heads) labeled by GFP-tubulin. The cyst of pre-meiotic cells is delineated by the dashed yellow line. (C) Cells in meiosis are identifiable by their large meiotic spindles that are easily visualized with GFP-tubulin. The cyst of meiotic cells is delineated by the dashed yellow line. Please click here to view a larger version of this figure.
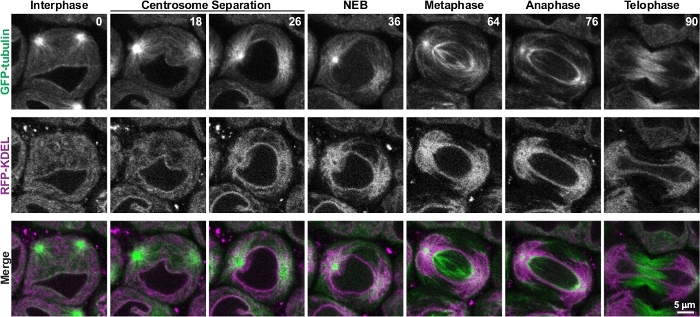
Figure 4: Time-lapse imaging of meiosis in a single spermatocyte. A single spermatocyte co-expressing GFP-tubulin and RFP targeted to the ER (RFP-KDEL) was imaged every two minutes through the course of meiosis. Shown are representative images of the spermatocyte at specific stages of meiosis, beginning in late interphase and progressing through telophase. Times are in minutes. Each image is a single optical plane from a stack of fifteen optical slices acquired at each time point. This figure was modified from Karabasheva and Smyth, 20198. Please click here to view a larger version of this figure.
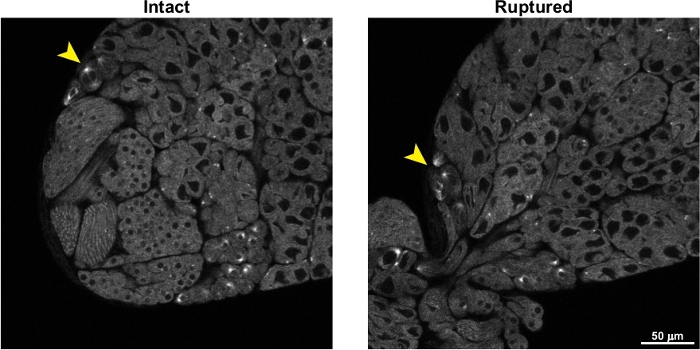
Figure 5: Representative images of a testis before and after rupturing. The image on the left, labeled “Intact”, shows a GFP-tubulin expressing testis just before rupturing. The image on the right shows the same testis after rupturing. Cysts of spermatocytes can be seen streaming out of the ruptured testis. The arrowheads indicate a cyst of meiotic spermatocytes; note the significant movement of these spermatocytes after the testis ruptures, making imaging these spermatocytes over time extremely challenging. Please click here to view a larger version of this figure.
Video 1: Full meiotic division of a single spermatocyte. Shown is the full time-lapse of the GFP-tubulin and RFP-KDEL expressing spermatocyte in Figure 4. Images were captured every two minutes, and the playback rate is three frames per second. This video was modified from Karabasheva and Smyth, 20198. Please click here to download this video.
Video 2: Ruptured testis. Time-lapse of a testis before and after rupturing. The rupture is evident due to the sudden streaming of cysts of cells through the lower left corner of the testis. Images were captured every two minutes, and the playback rate is three frames per second. Please click here to download this video.
