The animal procedures are performed following approved protocols of the National Institute of Health Guide for the Care and Use of Laboratory Animals. Approval to conduct the study is granted by the Animal Care and Ethics Committee (CICUAL) of INIMEC-CONICET-UNC (Resolution numbers 014/2017 B and 006/2017 A).
1. Primary rat cortical neuron cultures
- Anesthetize E18 Wistar pregnant rats in a CO2 chamber with a mixture of 80% CO2/20% O2 for 60 s of exposure and then sacrifice then by dislocate the cervical vertebrae.
- Perform the following steps in a laminar flow hood. Extract encephalon from the embryos with a forceps style # 3 and straight sharp small spring scissors (Table of Materials) to Hanks solution in cell culture dishes.
- Dissect the tissue under 20x magnifying glass, using two fine-tipped forceps style #5, separating the frontal cortex from the meninges. Transfer the frontal cortex to a 15 mL conical tube and incubate the tissue with 3-4 mL of Trypsin-EDTA (0.25%) for 15 min, at 37 °C for chemical digestion.
- After digestion, dilute it with 3-4mL of Dulbecco's modified eagle medium (DMEM) plus 10% fetal calf serum (FCS). Then wash it 3 times with Ca2+/Mg2+ free Hanks Balanced Salt solution and 0.1% glucose by centrifugation (3000 g for 5 min).
- Mechanically dissociate the tissue 10 times by pipetting up and down using a 1000 µL tip, in DMEM plus 10% FCS (Table of Materials). Then centrifuge the homogenate at 100 x g for 1 min, collect the supernatant and complete the remaining volume to 1000 µL.
- Plate the cell suspension on cell culture dishes at a density of 520 cell/mm2 with 2 mL of DMEM containing 10% FCS, 1% penicillin/streptomycin and 1% of an essential aminoacid additive (see Table of Material). Then incubate at 37 °C in a humidified environment with 5% CO2 for 2 h.
- Organic matter removal and poly-L-lysine coating of cell culture dishes
- Remove the organic matter by placing the glass material in a reaction chamber and add 96% ethyl alcohol to just about cover the material. Then add 68% (w/v) nitric acid drop by drop until brownish-greenish bubbles start to appear. Once this happens, partially cover the container and allow the reaction to continue until explosion ceases.
NOTE: Remove the organic matter under a gas extraction cabin to avoid exposure to noxious gases. - Before poly-L-lysine coating, rinse the glasses with sterile Mili-Q water about ten times. Then incubate the culture glasses and dishes with 0.1 μg/μL poly-L-lysine for 4 h before using them. After incubation, rinse the glasses with sterile Mili-Q water about ten times.
- Remove the organic matter by placing the glass material in a reaction chamber and add 96% ethyl alcohol to just about cover the material. Then add 68% (w/v) nitric acid drop by drop until brownish-greenish bubbles start to appear. Once this happens, partially cover the container and allow the reaction to continue until explosion ceases.
- Organic matter removal and poly-L-lysine coating of cell culture dishes
- To allow neural cell differentiation, change the medium for a serum-free neurobasal medium, with the serum-free supplement (Table of Materials). Keep cultures at 37 °C with 5% CO2 until treatment.
2. Lentivirus production
NOTE: The oligonucleotides containing the sequences targeting the 3'UTRs of either the CN-Aα isoform or CHOP and with the non-targeting sequences (scrambles) are listed in Table 1.
- For annealing, mix two single-stranded oligonucleotides with complementary sequences in equal molar amounts, using Annealing Buffer (10 mM Tris-HCl, pH 7.5-8.0, 50 mM NaCl, 1 mM EDTA).
- Heat at 95 °C for 2 min and then cool slowly by transferring samples from the heat block or water bath to a bench-top at room temperature. Dilute the resulting product [short hairpin RNA, (shRNA) with cohesive ends] to a final concentration of 0.5 µM, aliquot them and store at -20 °C.
- Clone the shRNA insert into lentiviral vector pLKO.3G, with GFP as a green fluorescent marker. For this, digest 1 µg of vector with EcoRI and PacI restriction enzymes by mixing the following in a sterile tube: 2 µL of 10x restriction enzyme buffer (100 mM bis-Tris propane-HCl, pH 6.5, 100 mM MgCl2, 1 mg/mL bovine serum albumin), 1 µL of 1 µg/µL pLKO.3G, 0.5 µL of 10 U/µL ECoRI, 0.5 µL of PacI (10 U/µL), and 16 µL of sterile ionized water.
- Mix gently by pipetting. Incubate the reaction for 3-4 h at 37 °C.
NOTE: Overnight digestions are generally unnecessary and may result in DNA degradation.
- Mix gently by pipetting. Incubate the reaction for 3-4 h at 37 °C.
- For ligation, mix the digested vector and insert in a molar ratio of 3:1 and incubate it with the T4 ligase and ligase buffer (provided by manufacturer) overnight at 16 °C.
NOTE: At this point ligation reaction can be stored at 4 °C until further use. - Transform DH5α competent cells (Table of Materials) or another suitable E. coli strain with the ligation reaction as follows.
- Mix competent E. coli with the total volume of ligation reaction and chill it on ice for 15 min, place it in a 42 °C bath for 2 min and then chill it on ice again for 15 min.
- Transfer the total volume subjected to heat shock to a 15 mL tube and complete the volume to 1000 µL with Luria-Bertani (LB) liquid medium that has been previously maintained at 37 °C. Shake the bacteria for 90 min at 37 °C in an orbital shaker at approximately 330 rpm.
- Centrifuge them at 5,000 x g for 5 min at 4 °C and discard 900 µL of supernatant. Use the remaining supernatant to resuspend the pellet. Spread bacteria suspension evenly over a solid ampicillin (100 µg/mL) LB plate.
- Incubate at 37 °C overnight, then check bacterial growth. At this point, the bacteria may be stored at 4 °C for 4-5 days.
- Pick a single colony to transfer into a 50 mL tube with 10 mL of LB liquid medium and ampicillin (100 µg/mL). Shake the bacteria ON at 37 °C at approximately 330 rpm.
- Centrifuge the culture to pellet the bacteria at 5,000 x g for 5 min at 4 °C, discard the supernatant and conserve the bacterial pellet. This can be stored at -20 °C.
- Purify the plasmid with a DNA purification kit from bacteria pellet following manufacturer's instructions (Table of Materials). Calculate the DNA concentration by measuring the absorbance at 260 nm and multiplying by the dilution factor, using the following relationship: A260 of 1.0 = 50 µg/mL pure double strand (ds) DNA.
3. Lentivirus infection
NOTE: Perform the lentivirus generation procedure in a Class II laminar flow hood.
- 24 h before transfection, seed 1-5 × 106 HEK 293 cells into 100 mm of diameter culture dishes in 8 mL of growth medium and incubate them at 37 °C, 5% CO2 overnight.
- Mix 14 µg of pKLO.3G vector with specific insert, 10.5 µg of packing plasmid psPAX and 3.5 µg of envelope plasmid pMD2.G (Table of Materials). Dilute 45 µL of plasmid transfection reagent (Table of Materials) in 700 µL of reduced serum media (Table of Materials) and incubate for 5 min at room temperature. Then combine the DNA mixture with the diluted plasmid transfection reagent, mix gently, and incubate for 20 min at room temperature.
- Meanwhile, change for fresh DMEM the medium of the culture dish of HEK 293 cell cultures (70% confluent at the time of transfection). Then add the entire volume of the DNA solution drop by drop. Rock the dish gently.It is not necessary to add/change medium after transfection.
- Incubate the cells at 37 °C, 5% CO2 for 48 h- 72 h to allow shRNAs to reach their optimum transduction checked by GFP fluorescence using a fluorescence microscope. Afterwards, collect the medium with lentivirus and store at 4 °C.
- Filtrate viral supernatant with a 0.45 µm nylon membrane. Aliquot the flow-through in 1 mL. Next, centrifuge at 17,000 x g for 4 h. Discard the supernatant and conserve the pellet. Allow invisible pellet to dry, and, once dried, store at – 80 °C.
- Perform viral titration by flux cytometry. Seed 4 x 105 HEK 293 cells in 24-well plates and incubate at 37 °C for 24 h. Then resuspend the viral particles in 200 µL of DMEM culture medium (Table of Materials) and add 0.05, 0.02, and 0.005 µL to each well.
- Incubate viral infection for 72 h, remove medium and trypsinize (0.05% trypsin, 1 mL) for 3 min. Add 1 mL of DMEM in 10% FCS solution and centrifuge at 500 x g for 5 min. Discard the supernatant and wash the pellet twice with PBS.
- Resuspend the pellet with 100 µL of PBS and fix the cells with 100 µL of 2% paraformaldehyde (PFA) in PBS. Analyze the cells with a flux cytometer and determine the percentage of GFP fluorescent cells. Collect this data to calculate viral titration using the following formula:
Viral Titration
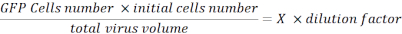
4. Primary neuron cultures stressed with ganglioside GM2 accumulation and immunocytochemistry using anti-MAP2 antibody
- Infect the primary cortical neuron cultures (15 days in vitro) with specific shRNA oligonucleotides targeting the 3' UTRs of either CN-Aα or CHOP and incubate them at 37 °C with 5% CO2 for 1 day.
NOTE: Primary cortical neuron cultures of 7-8 days in vitro could also be used if the cells show high neurite growth.- Prepare a 500 µM stock solution of ganglioside GM2 in ethanol and sonicate it for 1 h.
- Add GM2 stock solution to the culture dishes medium at a final concentration of 2 µM. Allow to incubate at 37 °C with 5% CO2 for 16, 24, and 48 h.
- Use some cultures to prepare cellular homogenate for western blot analysis (for antibody specification, see the Table of Materials).
- Follow steps 1.4. and 1.5. and then fix the cells with 4% PFA and 120 mM sucrose in PBS for 20 min at 37 °C, and then wash it with PBS. Perform this procedure on an anti-slipping membrane in a wet chamber. Afterwards, add permeabilization solution (0.2% Triton X-100 in PBS) for 5 min and wash with PBS.
- Use 5% BSA diluted in PBS for 45 min to allow blocking, then incubate with anti-MAP2 antibody, diluted 1:800 in blocking solution, at 4 °C overnight.
- At the end of the incubation time, wash twice with PBS and incubate with secondary antibody (Table of Materials) for 1 h. Then wash the cells with PBS and mount the glasses onto slides using an aqueous mounting medium (Table of Materials).
5. Neurite atrophy analysis
- Obtain images with a 20x air lens (NA 0.8) using an epifluorescence-inverted microscope equipped with a CCD-camera. Analyze images with ImageJ plug-ins. For this, load the picture to ImageJ and subtract the background by clicking on Process | Subtract Background.
- Click on Image | Adjust | Threshold or type the following keys: Ctrl (or Cmd) + Shift + T. A window pops open to adjust minimum values, allowing path and soma tracings to be distinguishable. Click on the binary image (neurites and somas are white colored). On this image, use freehand selection to delete somas; the selected soma area turns black again.
- Select traces and click Edit | Selection | Create Selection. Get the ROI by clicking Ctrl (or Cmd) and T keys of the selection and conserve it. Open the original image, then click on the ROI manager panel and select the belonging ROI, previously obtained. Then click on the original image.
- Once the ROI traces on the image, type Ctrl (or Cmd) and M keys (Analyze | Measure). A window pops open; copy the mean value (arbitrary units a.u.) to process statistical analysis.
NOTE: To obtain values as µm, click on Analyze | Set Scale. A window pops up open to set values. Be sure to add the correct scale (Analyze | Set Scale) depending on the characteristics of the microscope used.
Here, we address the question of whether silencing two PERK downstream components affects the transition phase of UPR in an ER stress cell model.To achieve this, we silence the CN-Aα gene as well as the CHOP gene by two specific shRNA sequences for each (Table 1) in primary neuron cell culture for 1 day10. The expression is analyzed by Western blotting (Figure 1 and Figure 2). A clear inhibition is observed of ER stress-mediated CN-Aα and CHOP increase in knockdown cells, but not in control cells (shRNA scrambles). It should be noted that the basal CN expression level is not affected with this treatment condition.
We also examine the possible link of GM2 accumulation-induced ER stress with neuronal degeneration by performing MAP2 immunostaining of primary neuronal cultures (Figure 3 and Figure 4).
Neurite atrophy is analyzed as total neurite outgrowth relative to the total cell number. This increases significantly after inducing ER stress by incubation of GM2 at 16-48 h. Interestingly, silencing of CN-Aα expression significantly enhances neurite atrophy, particularly at 16 h of GM2 accumulation, relative to GM2-untreated groups (Figure 3). Thus, CN-Aα knockdown accelerated the degeneration processes in neurons, corroborating the pro-survival effect of CN during the early phase of UPR. Conversely, CHOP knockdown resulted in significantly diminished neurite atrophy relative to controls, specifically at 16-24 h (Figure 4).
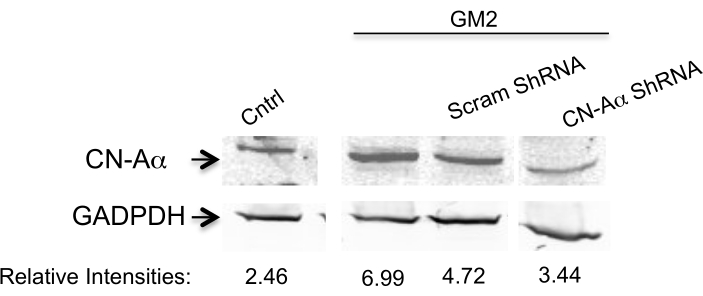
Figure 1. Data analysis for CN-Aα silencing efficiency. Knockdown is evaluated by Western blot analysis using antibody against CN-Aα and GADPH (loading control). The primary antibody is visualized by near-infrared fluorescence imaging system. Numbers below represent relative intensity ratio between CN-Aα and GADPH bands. This figure has been republished from Virgolini, M.J. et al.4. Please click here to view a larger version of this figure.
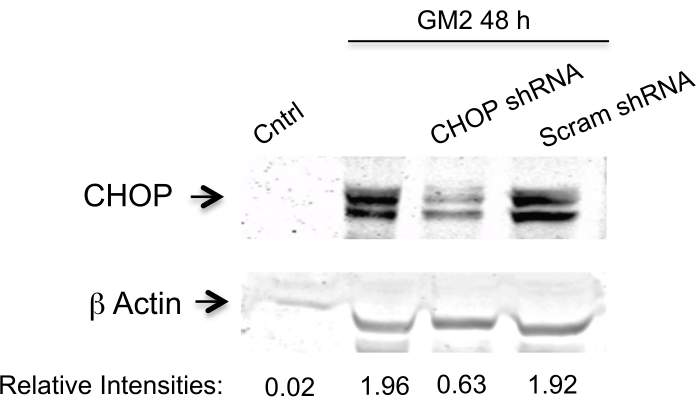
Figure 2. Data analysis for CHOP silencing efficiency. Knockdown is evaluated by Western blot analysis using an antibody against CHOP and β actin (loading control). The primary antibody is visualized and then the data are analyzed as indicated in Figure 1. This figure has been republished from Virgolini, M.J. et al.4. Please click here to view a larger version of this figure.
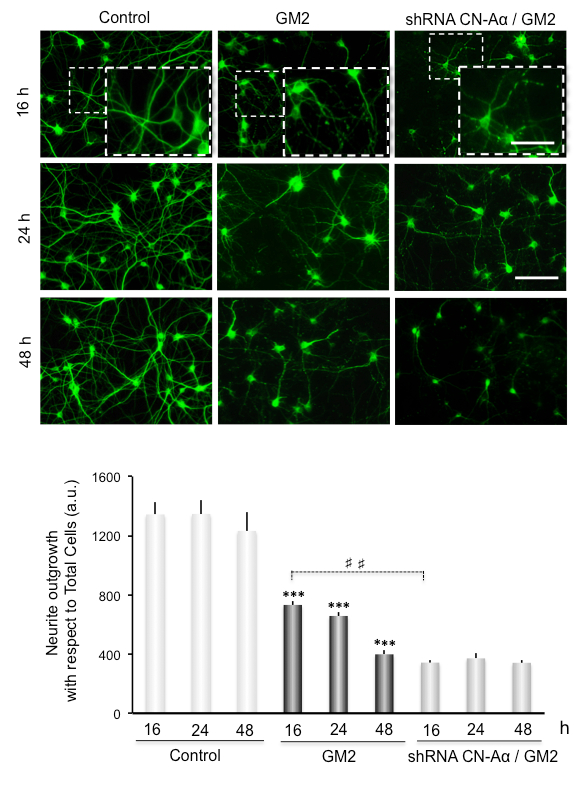
Figure 3. Data analysis for neuritic atrophy imaging in CN-Aα knockdown cells. (Top) Cultured primary neurons are loaded with GM2. Representative images of MAP2 immunostained cells are shown under control and experimental conditions. Images are recorded with an epifluorescence-inverted microscope equipped with a CCD-camera. Scale bars: 150 µm (regular images), 75 µm (magnified images). (Bottom) Histogram (mean ± SEM) represents neurite outgrowth with respect to total cells, analyzed by ImagJ plug-ins. *** and ###: p 0.0001 for comparison with the control, from one-way ANOVA. This figure has been republished from Virgolini, M.J. et al.4.
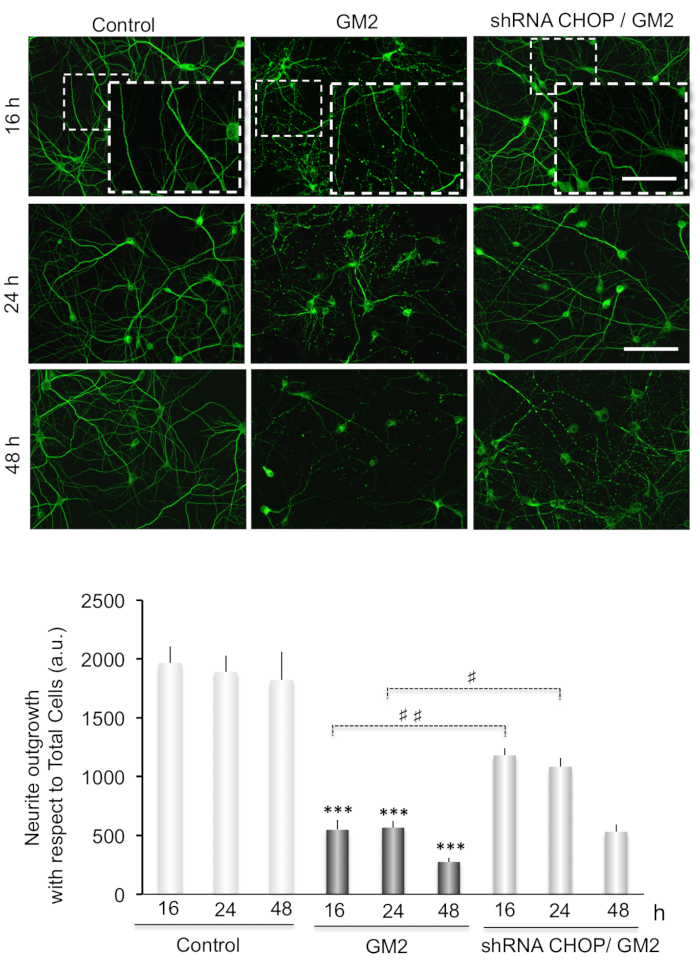
Figure 4. (Top) Data analysis for neuritic atrophy imaging in CHOP silencing cells. Primary neuron cultures are treated as indicated in Figure 3(top). (Bottom) Histograms and statistical notations as in Figure 3(Bottom) except ##: p≤ 0.001. This figure has been republished from Virgolini, M.J. et al.4. Please click here to view a larger version of this figure.
| Constructs | Sequence 5’ to 3’ shRNA |
| CN-Aα 1 | AATTGCCAGGAATTGGATTCAGTTTCTCGA GAAACTGAATCCAATTCCTGGCTTTTTTTAT |
| CN-Aα 2 | AATTCGCCAACCTTAACTCCATCAACTCGAG TTGATGGAGTTAAGGTTGGCGTTTTTTTAT |
| CN-Aα Scrb | AATTGAGTGAATTGTCGCTCTAAGTCTCGAG ACTTAGAGCGACAATTCACTCTTTTTTTAT |
| CHOP 1 | AATTGGTCCTGTCCTCAGATGAAATCTCGAG ATTTCATCTGAGGACAGGACCTTTTTTTAT |
| CHOP 2 | AATTTGAAGAGAACGAGCGGCTCAACTCGA GTTGAGCCGCTCGTTCTCTTCATTTTTTTAT |
| CHOP Scrb | AATTGAAGAGAGAAAGCGAACAATACTCGAG TATTGTTCGCTTTCTCTCTTCTTTTTTTAT |
Table 1. Forward construct sequences inserted to pKLG0.3 viral vector. Scrb: scramble.
