The basic protocol for in utero electroporation has been described in detail in another JoVE article from the Kriegstein lab 1, 2. This technique was originally described in the Osumi lab 3 and our protocol is based upon one developed in the LoTurco lab 4. We will provide an overview of the our protocol for in utero electroporation of rat embryos, focusing on the most important details, followed by a description of our protocol that applies in utero electroporation to the study of gene function in neuronal process outgrowth.
1. In utero Electroporation
- Preparing DNA and loading needles
The first step for in utero electroporations is to design your experiment to determine what DNA constructs you want to inject. This method is useful for both misexpressing or knocking down genes of interest. If you are planning on misexpressing or overexpressing a gene, be sure to use a promoter that is active in neuronal precursor cells. We recommend the CAGGS promoter, which consists of the chicken beta-actin promoter and the CMV enhancer 5. Since only a small subset of cells are transfected using in utero electroporation, it is critical to include a plasmid encoding a fluorescent protein such as GFP so that you can follow those cells that were successfully electroporated. For the plasmid encoding GFP, we recommend preparing the DNA at a concentration of 0.5 μg per microliter. For shRNA constructs, we have found that 0.5-1.0 μG per μL results in efficient knock down of your gene of interest. For overexpression or misexpression, we use between 1.0 and 3.0 μG per microliter, depending upon the size of the gene and the level of expression that the experiment calls for. DNAs are prepared using a Qiagen endotoxin-free prep kit, and diluted in 1 x PBS. We inject approximately 0.5-1.0 μL per embryonic brain, so, for a litter of animals we prepare 10 μL of DNA mixture for injection. We add 1 μL of Fast Green to the DNA so that we can follow the injected DNA.
Pulling needles to the correct shape is a critical step. Walantus et al. uses a different system for delivering the DNA and so their needle prep is also slighly different then ours1,2. The settings that you use to pull your needles will depend upon the brand of needle puller that you have. We use Model 750 from David Kopf. The settings we use are : Heat 1: 9.0, Heat 2: 0, Soleniod: 0, Filament size 3.0 mm, Heater Proximity: 3 mm, Time: 10 sec. Once pulled, we cut our needles with a razor blade at a ~45 degree angle such that the distance from the largest part of the opening to the tip is 11 mm. We then load the DNA from the back end of the needle. We then fill the remaining space in the needle with corn oil. For DNA injection, we use a Picospritzer III. Depending upon the exact bevel that is cut for each needle, we set the Picospritzer from 4.0 to 6.0. We use a foot pedal to deliver the pressurized air that expels the DNA from the needle. - Preparing animals for surgery
We use pathogen-free Sprague Dawley rats exclusively for these surgeries. Several other labs use mice of varying genotypes as well. Here, we describe show our protocol for electroporation of E15 rat embryos, but in utero electroporation is routinely performed in rats between the ages of E13 and E18. While early stage electroporation targets deep layers of the cortex, later stage electroporations target more superficial layers.
Animals are given a pre-operative dose of buprenorphine (0.05-0.1 mg/kg) before the surgery starts. There are multiple options for anesthetizing the animal. Walantus et al. utilizes isofuorane inhalation, while we routinely use intraperitoneal injection of ketamine (40-80 mg/kg) and xylazine (5-10 mg/kg)1,2. A toe pinch should always be performed to ensure that animals are fully anesthetized and unresponsive. Animals are kept on a heated pad throughout the surgical procedure.
The animal’s fur is shaved in the region of incision, and washed three times with ethanol followed by three times with iodine. An incision is made in the skin just lateral to the midline, followed by an incision in the muscle. The uterine horns are exposed very carefully. They are gently teased out of the body cavity using your fingertips. Keep embryos hydrated with sterile PBS while they are outside of the body cavity. - Injecting DNA and electroporation
When you first start performing these surgeries, the hardest part is becoming familiar with where you need to inject the DNA so that you fill the lateral ventricles, and getting used to how deep you inject your needle in order to hit the correct region. Embryos are gently manipulated with your fingertips so that you can identify where the head is, and if you look closely you will be able to see the midline suture. This serves as a general landmark that you can use to determine where the lateral ventricle is located. We inject the DNA through the uterine wall and into the lateral ventricle. We use a footpedal to control the injection of the DNA – multiple pulses of DNA are performed until the lateral ventricle is filled with the DNA/dye mixture. We then place paddle electrodes on either side of the head of the embryo and use another footpedal to deliver the pulse across the head of the embryo. The placement of the electrodes is critical in determining which region of the cortex is electroporated. Since DNA is negatively charged, the DNA will travel toward the positive electrode when a charge is dispelled across the paddles. Depending on the exact placement of the electrodes, different subsets of cells will be targeted. We routinely place the positive electrode near the dorsal-medial positions across the cerebrum. However, the LoTurco lab beautifully showed that if you place the electrodes in more ventral lateral regions of the cerebrum you can target the cells of the cortical-striatal boundary and hit cells of the lateral cortical stream 6. Each embryo can be electroporated, and different combinations of DNA constructs can be used in each embryo. - Suturing and post-operative care
Following electroporation of all embryos, the uterine horns are carefully returned to the body cavity, and both the muscle layer and the skin are sutured. The technique for this is outlined in Walantus et al1,2. Animals are monitored continuously until they recover from anesthesia, and the analgesic buprenorphine (0.05-0.1 mg/kg) is administered every 8-12 hours.
2. Culturing Electroporated Cortical Neurons
- Harvesting electroporated brains and dissecting electroporated region
For in vivo analyses following in utero electroporation, animals can be harvested at any time point from 24 hours following electroporation to early after birth to adulthood. However, when culturing primary neurons we harvest 24 hours following electroporation at E16. At this time, the electroporated embryos are expressing detectable levels of GFP.
Animals are euthanized using carbon dioxide inhalation and rapid decapitation. Embryos are dissected out of the uterus and placed in HBSS with divalent cations, keeping track of which embryos were electroporated with which DNA plasmids. It is critical to use filtered HBSS, sterile tubes and plates, and autoclaved tools for dissection. The cortices are dissected out and the meninges removed using a microscope in a hood. These cortices are then observed under a dissecting microscope with the capacity to visualize GFP. GFP positive regions of the cortex are identified, and we ue a pair of vanna scissors to cut out the GFP positive regions from the cortex. These pieces are placed in HBSS without divalent cations in a 15 ml conical tube. - Dissociating and plating neurons
Once all GFP positive regions are dissected, HBSS is replaced with 0.25% trypsin, and incubated at 37 degrees for 5 minutes. Trypsin is removed, replaced with plating media (DMEM + 5% FBS + Penn/strep + glutamine), and triturated 5-7 times to dissociate the cells. Volumes used depend upon the amount of tissue present. Dissociated cells are then plated on CC2 coated chamber slides. For two chamber slides, we plate 200,000-350,000 cells per chamber in a volume of 1.5 mL of plating media. After 4 hours, plating media is aspirated and replaced with 1.5 mL Neuronal culture media (Neurobasal media + B27 supplement + glutamax + gentamycin) per chamber.
3. Analyzing Neuronal Process Outgrowth
- Fixing and immunostaining cultures
In order to measure short term effecs of genetic manipulation of these cells, we harvest the primary neurons after three days in vitro. If primary neurons are to be cultured longer for additional analyses, half of the media should be replaced every three days. For fixing cultures, we aspirate the media from the chambers, and fix the neurons in 4% paraformaldhyde for 15 minutes. Following fixation, cells are washed two times in PBS and then put in blocking solution (2% donkey serum with 0.1% Triton X-100 in PBS) for one hour. Cells are then incubated in primary antibody for 1 hour. For analyses of neuronal process outgrowth, we use anti-beta tubululin antibody to identify neurons – beta II tubulin immunostaining labels the neruonal cell body, dendrites and axons. Cells are then washed three times in PBS for five minutes, and then incubated in Cy3-anti mouse for 1 hour, followed by three more PBS washes, counterstaining nuclei with DAPI and mounting. - Measuring neurite length
Images of GFP positive, beta-III tubulin positive neurons are acquired on a Zeiss Axioskop with a MC100 camera system. Several variables can be examined in these GFP positive cells including length of all neurites, length of the longest neurite, branching of neurites, size of cell soma, etc. We have used this method to analyze neuronal process outgrowth upon knock down or overexpression of genes relating to neurodevelopment and neurodegeneration, with a focus on neuronal process length. In order to measure neuronal processes outgrowth, we use Axiovision LE 4.4 software (from Zeiss). Within this software, there is an option to select the “outline” tool. Using this tool, you can use your computer’s mouse to trace the length of each neuronal process. It is critical to define the objective that you are using in order to get an accurate measure of your neurites. For these analyses, we usually use a 20x objective.
4. Representative Results
We have found that Sprague Dawley litter size ranges between 6 and 14 embryos. We usually electroporate all of the embryos. Each embryo can be electroporated with a different combination of DNAs. However, we usually electroporate at least four brains with the same condition and pool these brains before dissociating and plating.
We have found that with this technique approximately 75% of electroporated brains are targeted to the desired region of the cortex, whether that be dorsal medial or ventral lateral cortex (Figure 1). In addition, we have found early electroporations at E13-14 target deep layer neurons such as Tbr1 positive layer VI neurons, while later electroporations target CTIP2 positive, TBR1 negative layer V cells, and still later electroporations target Brn2 positive layer II/III cells. An excellent description of different markers and explanation of neuronal subtype specification in the cortex in found an article by Moleneaux et al 7. Figure 2 shows coronal sections of brains electroporated at either embryonic day 15.5 or 17.5 and harvested at postnatal day 5. Shown in red is immunostaining for Oct6. You can immunostain for markers in culture to confirm what cell layer populations you have targeted. We have found that you can expect to target the same cell layer population of cells in every embryo of the same litter (in other words, the targeting depends upon the embryonic timepoint rather then on other technical variations).
In culture, the percent of cells that are GFP positive can range widely depending on how conservative you are when dissecting out the GFP positive region (Figure 3). However, even when we are very conservative and dissect out only the GFP positive patch of cells, the highest percentage that we observe is 5-10% – although you are dissecting the region of the cortex that was electroporated, cells in only one layer will be targeted. This low transfection effciency is helpful in identifying which processes belong to the electoporated cell that you are analyzing. Plating cells at this higher density contributes to having heathier cultures, however, it is difficult to discern which process belongs to which cell body in the GFP negative cells (Figure 3).
If you have trouble seeing all of the fine processes of the electroporated cells, you can either increase the concentration of GFP DNA that you are electroporating to increase expression of GFP, or you can immunostain the dissociated cells using an anti-GFP antibody (from Invitrogen) along with a Cy2 secondary antibody.
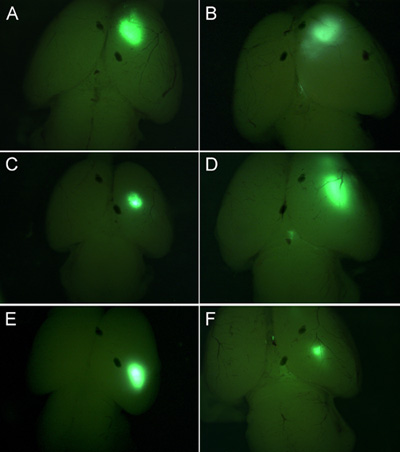
Figure 1. E15.5 Sprague-Dawley rats were electroporated with GFP plasmid and harvested three days later. Based upon the placement of the electrodes, different regions of the cortex will be targeted. A-F show GFP fluorescence in whole brains.
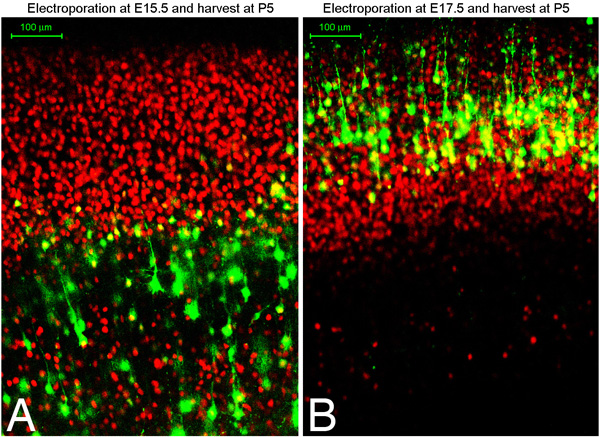
Figure 2. E15.5 (A) or E17.5 (B) Sprague-Dawley rats were electroporated with GFP plasmid and harvest at postnatal day 5. Brains were fixed, sectioned coronally using a vibratome (100 micron sections), and immunostained for Oct6 (red). A and B show confocal images of immunostained sections.
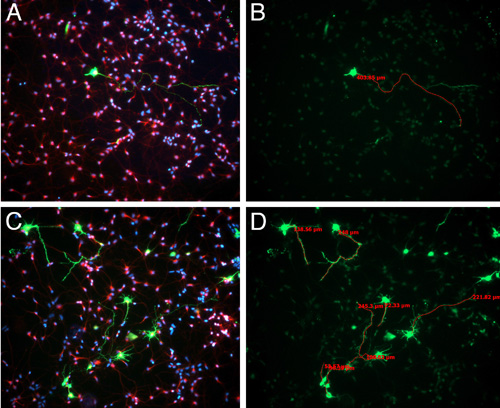
Figure 3. E15.5 Sprague-Dawley rats were electroporated with GFP plasmid. 24 hours following electroporation, brains were harvested and GFP-positive, electroporated regions were dissected and dissociated, as described in the video. After 3 days in vitro, cells were fixed and immunostained for bIII-tubulin (red) and staining nuclei with DAPI (blue) (A,C). The length of the longest neurite was measured using Axiovision software (B,D).
