自己組織化反応押出、マイクロフルイディクス, 経由で複数の長さスケールでの分解性感温ハイドロゲルを加工・ エレクトロスピニング
Summary
分解性感温ハイドロゲル ヒドラゾン架橋高分子オリゴマー バルク スケール、マイクロ スケール、およびナノスケール、ゲル粒子・ ナノファイバーの調製のための後者のに基づいて作製のためのプロトコルを説明します。
Abstract
その究極の臨床使用を生物学的関連性の欠如によって妨げられている様々 なスマート材料は、さまざまな生物医学アプリケーション (例えばドラッグデリバリー、ティッシュ エンジニア リング、バイオ イメージングなど) の検討されている間最もスマート材料の観察の劣化。特に、ほぼ一様に、機能的分解性ポリマーに基づく温度応答性ハイドロゲルのです (例えばpoly(N-isopropylacrylamide) (PNIPAM) または (oligoethylene リコール ポリメタクリル酸 (POEGMA)).可変セル材料相互作用、潜在的な theranostic 材料細胞骨格のリモート制御や代謝規制薬物送達の課題に感温ハイドロゲルの可能性を効果的に翻訳するなど、イメージングとドラッグデリバリー システムと他のアプリケーションのメソッドは (完全分解ではない) の場合、少なくとも腎クリアランスの材料の必要な有効期間を次のことができる、ヒドロゲルをレンダリングに必要なです。そのために、このプロトコルはヒドラジドと分子のアルデヒド官能基化 PNIPAM または POEGMA オリゴマーの反応に基づいて複数の長さスケールで加水分解分解ヒドラゾン架橋ハイドロゲルの調製をについて説明します。腎の濾過限度以下の重み。具体的には、分解性感温一括ヒドロゲル (ダブル バレル シリンジ法を使用して) を作製する方法ヒドロゲル粒子 (同時混合を促進するマイクロ流体プラットフォームを使用して両方のマイクロ スケールで、前駆体ポリマーの熱対流を使用してナノ乳化自己架橋法と)、ハイドロゲル ナノファイバー (反応性エレクトロスピニング戦略を使用して) 記述されています。それぞれのケースで従来自由ラジカル架橋プロセスを介して達成に類似した温度応答性のプロパティを持つハイドロゲルを達成することができます、しかし、ヒドラゾン架橋ネットワークは、オリゴマーに再編成する時間の経過と共に低下する可能性が前駆体高分子と有効にするクリアランス。そのため、(これが一般的、合成に応用水溶性ポリマー、スマート材料なだけではなく) これらのメソッドは合成スマート材料臨床応用への移行が容易になります期待しています。
Introduction
スマート材料は、外部および/または環境の信号にリバーシブルの「オンデマンド」応答のための潜在性ための重要な注目を集めています。温度応答性材料は温度 T での沈殿物の温度駆動の結果下の臨界溶液温度 (LCST) 動作により特定の関心を集めている > LCST1,2。感温ハイドロゲルのコンテキストでこの下限臨界溶液温度現象は可逆 swelling/腫れイベント一括温度可変サイズの結果によって明示される (T で大きく < LCST)3細孔のサイズ (大きい T で< LCST)4、および界面特性 (T で親水性 < LCST)5。このような遷移は、ドラッグデリバリーに広く適用されている (外部または環境にトリガー可能な薬物4,6,7をリリース)、組織エンジニア リングと細胞培養 (可逆細胞粘着のため/はく離8,9,10)、(切替可能な膜の気孔率と浸透率またはサーマル リサイクル可能の診断サポート11,12,の版13)、マイクロ流体処理 (オン ・ オフ弁の流れ14,15を調整する)、およびレオロジー修飾子 (の温度可変粘性16)。重要な (そして増加) 仕事も oligoethylene リコール (ポリメタクリル酸に行ったものの、最も一般的感温ハイドロゲルは、poly(N-isopropylacrylamide) (PNIPAM)17、に基づいてを調査 (POEGMA)2 ,18 , poly(vinylcaprolactam) (PVCL)19,20。POEGMA は、数の異なる基を有するモノマーの線形予測を混合物の21,22の予想される改善された生体適合性とその曲安易な LCST 挙動を与えられた特定の最近の関心を集めています。エチレンオキシドの側鎖の繰り返し単位を変えたり ~ 20 ° C から LCST > 90 ° C2,23。ただし、これらのポリマーのラジカル重合により作製した、従って潜在的なユーティリティおよび生物医学アプリケーションのコンテキストでこのような高分子の訳しを著しく抑える炭素-炭素バックボーンが含まれています分解 (または少なくとも腎のろ過によりクリアランスのための容量) の要件は、通常。
この制限に対し、我々 が最近広く報告ヒドラゾン化学の応用で (すなわち、ヒドラジドとアルデヒド官能基化前ポリマー反応) 感温の分解性類縁体を準備するには。ヒドロゲル24,25,26,27,28,29。機能性前駆体高分子30の混合時に酸ヒドラジドとアルデヒド グループ間急速かつ可逆的反応により両方の in situゲル化 (安易な注入を必要とせずにこれらの材料を有効にする手術注入または任意の種類の UV 照射や化学開始など外部重合刺激) 化学架橋サイトの密度によって制御される速度でネットワークの加水分解の分解だけでなく。さらに、腎の濾過限度以下ヒドロゲルを準備するために使用前のポリマーの分子量を図り、このアプローチを使用したゲルは、25 本体から消去することができますオリゴマー前駆体ポリマーに戻って低下します。 ,,2728。低毒性とこれら材料25,26,27による低の炎症性組織反応と相まって、このアプローチを提供する可能性のある変換可能な感温の使用法医学、特に場合は、すべての長さにこのようなヒドロゲルの制御された分解性アナログ スケール (一括、マイクロ、ナノ) スマート ゲルを作製できます。
このプロトコル合成感温前ポリマー ヒドラジドとアルデヒド グループと同様の方法これらのポリマーに適用する上明確に定義された寸法を持つゲルを作成する制御番号、機能集積化を行うための方法について述べる様々 な長さのスケール。特に、この原稿は、反応性酸ヒドラジドとアルデヒド官能基化前ポリマーの混合を制御する開発し、, このように明確に定義されたジオメトリと感温ハイドロゲル ネットワークを作成する 4 つの異なるアプローチを説明しますと形態:
スタティック ミキサーをそのコンセントに装備して、その後に共押出ダブルバレル シリンジの樽に反応前のポリマーを読み込まテンプレート戦略を説明、定義されたサイズに分解性一括ヒドロゲルを作成する、シリコン金型目的ハイドロゲル形状および寸法21,27 (図 1)。
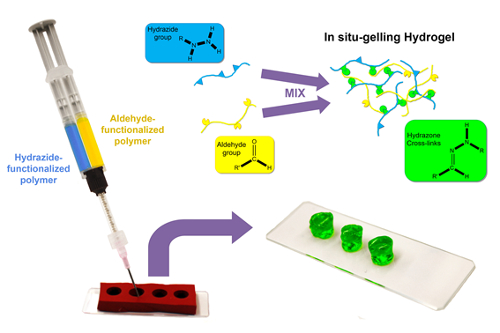
図 1: 一括ハイドロゲル形成の模式。ヒドラジドとアルデヒド官能基化高分子溶液 (水または緩衝水溶液) はダブル バレル シリンジの樽に読み込まれ、円筒形のシリコーン型に、このスタティック ミキサーを通して共押出します。ミックス フォーム ヒドラゾン架橋ハイドロゲルが自立 (一度金型を削除) を分前駆体高分子の濃度および官能基密度によって数秒以内にその場で急速なゲル。この図の拡大版を表示するのにはここをクリックしてください。
ミクロン スケールの分解性ゲル粒子を作成するため反応マイクロ メソッドが記載されて、前駆体高分子溶液は、同時に混合し、ソフト リソグラフィ テンプレート マイクロ流体チップ設計を使用して、有効にすることを乳化、混合反応性高分子液滴の形成その後ゲルその場でテンプレートのサイズとフォーム ゲル微粒子乳剤 (図 2)31,32。
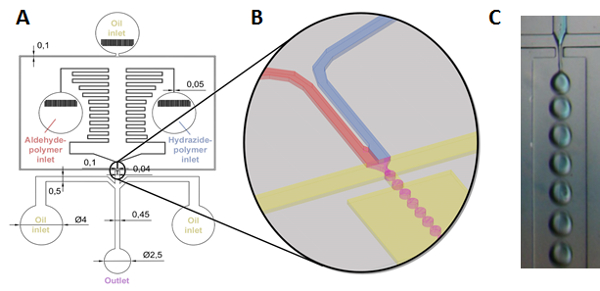
図 2: ゲル微粒子形成反応マイクロ流体を介してのスケマティック。(A, B)ヒドラジドとアルデヒド官能基化高分子溶液 (水または緩衝水溶液) は、ジグザグの一連の水路の逆流を防いで圧力勾配を作成するように設計を介して下流に接続されている別の貯水池にシリンジ ポンプによって供給されます。ポリマー、(またシリンジ ポンプによって駆動される) 両側から流れるパラフィン オイルでせん断される直前に混合し、水の流れ中心生産の結果ノズルを通して強制連続パラフィン油相 (高分子溶液) 液滴(ノズル面積と液滴形成過程の図の (B) 参照)。追加の 2 つパラフィン油入口ノズルをさらに別のコレクション チャネル流、その後生成される微粒子ゲル粒子除去する前に完全なゲル化への水滴の後配置します。磁気撹拌ビーカー; で収集(C) ノズルの液滴生成プロセスの画像 (そのヒドラジド ポリマーは混合を説明するために青というレッテルに注意)
反応前駆体ポリマー (「シード」高分子) の 1 つのソリューションがあり、安定した nanoaggregate を形成するための LCST 上記方法について説明自己反応性熱駆動、ナノスケールの分解性ゲル粒子を作成するには相補的な反応前駆体ポリマー (「架橋」ポリマー); 添加による架橋後結果ヒドラゾン架橋ナノゲル28nanoaggregate (図 3) で直接テンプレート化されたサイズがあります。
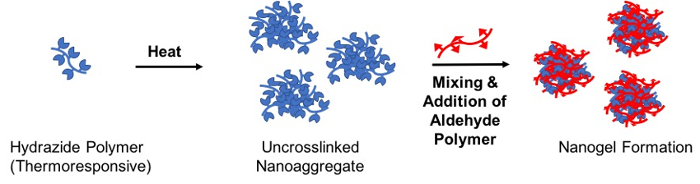
図 3: 熱駆動を介してナノゲル形成反応の概略図自己集合。上記の安定した uncrosslinked nanoaggregate を作成するその下限臨界溶液温度 (感温) ヒドラジド官能基化ポリマーを含む水溶液を加熱します。次、アルデヒド官能基化ポリマーはヒドラゾン結合形成を介して nanoaggregate 架橋に追加し、したがって、LCST 以下で冷却にナノゲル粒子を安定させます。この図の拡大版を表示するのにはここをクリックしてください。
(一括ゲルを作るために使用)、そのコンセントでスタティック ミキサー搭載ダブル バレル注射器は標準エレクトロスピニング プラットフォーム (図 4 に接続されている反応エレクトロスピニング法を説明分解性ナノファイバーの作製、)33。
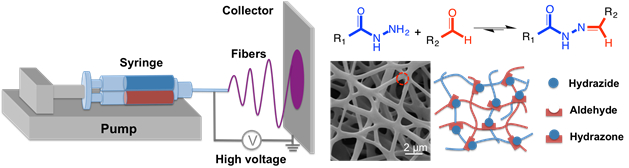
図 4: ハイドロゲル ナノファイバー形成反応エレクトロスピニング模式。スタティック ミキサー (一括ヒドロゲルのとおりロードしますが、またエレクトロスピニングの援助として高分子量 poly(ethylene oxide) の分数を含む) でダブル バレル注射器が接続されている注射器の終わりに針とシリンジ ポンプにマウントされています。高電圧電源。ヒドラゾンの架橋は、ストリーム (アルミ箔やアルミ円板) コレクターに達するナノ形態を維持するため、紡糸プロセス中に発生します。この図の拡大版を表示するのにはここをクリックしてください。
興味のポリマーとして PNIPAM または POEGMA のいずれかを使用してこのプロトコルで分解スマート ハイドロゲル ネットワークを作成するこのような手法の応用を実証します。粘度の適切な調整はあるものの、任意の水溶性ポリマーに記載されている基本的なアプローチを翻訳ことができますただし、(の場合、自己組織化ナノゲル作製法) 種子の形成の前のポリマーの安定性nanoaggregate。
Protocol
Representative Results
Discussion
PNIPAM と POEGMA; 上記の詳細で説明する方法の微妙な違いだけを使用して複数のポリマー システムにこれらのすべての加工技術を適用した正常に我々ただし、これらのプロトコルのユーザーは、これらのプロセスに他のポリマーに置換する際に生じる可能性のある潜在的な問題を認識しなければなりません。特に、2 前駆体ポリマーの混合の効率と同様、両方の加工性 (特にマイクロ法) で、前駆体ポリマーの粘度を増加することができます悪影響。さらに、高分子のゲル化時間は流れを阻害、または目的を形成する不可欠な反応前ポリマーの拡散を防ぐため、早期のゲル化を避けるためにターゲット形態に依存率で制御する必要があります。均質ゲル構造。それぞれの戦略だけでなく、我々 は各加工長さスケールでこのような制限に対処するためのこれらのアプローチに適応する使用しているアプローチの具体的な制限は次のとおりです。
ダブル バレル注射器共押出によるヒドロゲルを一括します。
ゲル化時間一括ヒドロゲルを形成するためダブル バレル シリンジ法の有効性を確保するためのコントロールにキーの変数です。お問い合わせの時にあまりにも速くゲル高分子 ( 5 s が望ましい (必須でない) がこのテクニックを使用するためこれは各ハイドロゲル キャストが同じ組成を持っていることを確認する物理的または機械的分析のため複製ヒドロゲルをキャストされている場合に特に重要です。ゲル化時間は 1 つの反応性官能基の密度を変更することによって簡単に変更できますまたは前駆体ポリマー (遅いゲル化につながる官能基密度が低い) または前駆体高分子の濃度を変更することの両方を使用する形式のゲル (遅いゲル化につながる濃度を下げる)21。代わりに、結果ハイドロゲル35 の構成を大幅に変更せずゲル化時間が短縮されます (反応が弱い) ケトン基とゲル化のペアの求電子剤として大幅 (反応) のアルデヒド グループを置き換える;ポリマーは、アルデヒドの混合調製、使用される前駆体高分子の濃度 (そして形成されたゲル中の固形物の量の割合) を変更することがなく必要に応じてゲル化時間をチューニングするケトン単量体前駆体を使用できます。
また、最初のハイドロゲル キャストがないこと常に後続ヒドロゲル、キャストで 2 つのバレルの内容は実際にスタティック ミキサーを到達速度の差に起因する観察と同じプロパティを注意すると思います。通常にダブル バレル注射器を押し出して小さなプライム結果として (< 0.3 mL) そのような変動を最小限に抑えるために、鋳造を開始する前にゲルのほんの一部。最後に中通常問題オリゴマーの合成前の高分子を使用する場合、1 つまたは複数の前駆体高分子溶液の粘度は両方の親指シンプルなうつ病を使用してフローを促進する観点から、この技術のコンテキストで挑戦を提起することができます。効果的なスタティック ミキサー内で混合を促進します。ただし、多少、驚くこともはっきりと異なる粘度による高分子溶液の前駆体まだ形成比較的均質ゲルのパーツリストに記載されているスタティック ミキサーの添付ファイルの使用 (例えばPNIPAM 高分子重量炭水化物26)、粘度のミスマッチの結果として非効率的な混合について懸念を示唆でないかもしれない重要な少なくともバルク スケール。必要な場合、ドライブの流れに (親指) の代わりにシリンジ ポンプの使用および/または出口より大きいゲージの針の使用がこれらのシステムで、押出し加工性に関連する問題を克服する助けることができます。
反応性マイクロ流体を介してマイクロ ヒドロゲル
ゲル微粒子の作製のためのマイクロ流体アプローチに関連付けられている重要な 2 つの反応性高分子マイクロ流路チップの起爆剤です。差圧が貯水池に 1 前駆体高分子溶液の逆流を運転できる場合はポリマーは、チップにさまざまな圧力または異なるレートで配信される、(または少なくとも貯水池に向かって) 他の前駆体ポリマーの。これは、結果、上流をゲル化、粒子の形成の流れを効果的に遮断、したがって切りくず処理を必要とするから。各貯水池とミキシングのポイント捺印拷問パス逆流; に大きな抵抗を作成しますただし、安定供給体制を実現するためにも訓練を受けたオペレーターはチップをゲル時折。我々 の経験を基に、1-2 分間 (時間をかけて比較的多ゲル微粒子が生成されます)。 液滴形成の開始に続く流れを安定させるために通常必要です。操作の最初の 5 〜 10 分内で問題が発生しないこと場合、は、連続単分散粒子生産のいくつかの時間が得られることが考えられます。非瞬時ゲル化時間と同様、比較的よく一致した粘度と前駆体ポリマーの使用 (少なくとも > 15 s が望ましい) でこのような問題を回避し、安定したフローの形成の推進を支援します。
メモこと、0.01 0.1 に至る様々 な流量 mL 水相中に h と油相に 1.1 5.5 mL/h は 25 〜 100 のサイズの範囲の粒子の作製につながる、このチップの設計を使用してテストされている/μ m で適用せん断によると、ジャンクションの流れ中心;高いせん断速度で流れを同一視して31,32に形成される小さい粒子。総水の流れの率が低い (~0.03 mL/h、プロトコルに引用として) が状単分散またはデバイスの寿命を損なうことがなくゲル微粒子のサイズを制御する最も効率的なことが判明した保っている間オイル流量を変化させ、どちらが引用全水系流量のより高い終わりを大幅に削減することが観察。大きな油流量 (> 5.5 mL/h) 小さい粒子を作成する可能チップはく離 (プラズマ接合 PDMS マイクロ流体チップで発生した一般的な制限) のリスクを増加させるが。別の方法を使用してチップを接着流れの速度でおよびこうしてより小さいゲル微粒子の生産、探っています現在戦略を有効にします。ノズルのサイズを小さく粒子を形成する前の早期のゲル化の高まりリスクではある生成可能性が微粒子のサイズを小さくことができます。流れの不安定性とこうしてより高い polydispersities とチップのゲル化; のリスクの増加につながる傾向が低速の流量高い安定性とこのプロトコルで使用される標準的なシリンジ ポンプより高い解像度を持つマルチ チャンネルのマイクロ フロー コントロール システムを使用してこの制限を克服できます。
オイルの選択は重いオイル (コレクション後ゲル微粒子の凝集を予防する上で有利な) としてこのプロトコルの成功にとって重要で報告された光シリコン オイルよりもノズルで大いにより少なく一貫した粒子の形成につながったプロトコル。我々 はこの減少を仮説再現性混合時点より変数せん断につながる重いオイルのシリンジ ポンプの一貫性は低くの結果であります。挑戦、特にすぐにその時点でその場でゲル化はなかった完全な大きい数字の反応マイクロ流体デバイスからの出口もあったコレクション フラスコでゲル微粒子の凝集を避ける機能グループは、フォーム コレクション バスに衝突する粒子の間に橋が利用できました。によりこのような課題が解決される: 増加するマイクロ流体チップ自体に出口チャネルの長さ、; より完全なゲル化を促進するために時間の長い期間のため層流ゲル微粒子を維持ノズル自体または粒子生産率; せん断場に影響を与えずにこのポスト混合チャネルでゲル微粒子チップとこより良い個別に多くの石油を供給するノズルの後側のチャンネルを追加します。ゲル微粒子の沈降を避けるためにコレクション フラスコに磁気ミキサーを追加し、隣接粒子間のより大きい平均分離を維持します。非常に遅いゲル化ポリマーだろう可能性が高いデバイスの安定性を向上、プライミングの問題を最小限に抑えるため、このようなシステムも観察された反応機能グループの大きい数としてのゲル微粒子凝集の危険性を大幅に向上未反応 (およびこうしてフォーム粒子間橋こと) のまま時間の長い期間にわたって。ゲル化時間 15-60 s の順序がこの手法に最適のように見えるよう、: 遅いプライミングが十分に速くほとんど反応性官能基を確実に有効にするのには十分に流チャネルを終了するゲル微粒子の前に消費される、コレクションのフラスコ。
最後に、テンプレートの油の除去は、結果として得られる粒子のスマート プロパティは追加前の高分子の組成に基づいて予想される維持し、生体のコンテキストでこれらの粒子の使用を有効にするのに不可欠です。洗浄手順ペンタンは一般的なゲル微粒子の生産のためにこの点で非常に効果的だった。ただし、直接生体文脈 (例えば、オンチップ細胞のカプセル化) でこの技術のアプリケーションには、このプロトコルの再評価が必要となります。連絡のコンテキストでより不活性油細胞36、分散剤として示唆されたオリーブ オイルの使用もしてきました。粒子の形成は可能でしたが、ゲル微粒子の集団は現在のチップ設計と、少なくとも鉱物油で達成できるよりもより多くの多を有意.したがって、チップは、合成高分子、天然高分子ゲル微粒子形成31の両方に適応するのに見えますが、変更されたデザインは材料のすべての組み合わせの間でより広くこのテクニックを悪用する必要があります。
ナノスケール ゲル反応による自己集合
シード高分子濃度の異なるを含む条件の処理の非常に広い範囲を使用して形成されたナノゲル (0.5-2 wt %)、crosslinking:seed 高分子 (0.05-0.2)、異なる温度 (40-80 ° C)、異なる混合速度 (比が異なる200-800 rpm)、およびさらに架橋ポリマー (2 60 分)28回異なる暖房。濃度、傾向は crosslinker:seed ポリマーのより高い比率の架橋密度は高くナノゲルへつながるし、低くなる種子高分子の高濃度を大きくナノゲルへリードとして予測が一般的にthermoresponsivities。それは高すぎる濃度は最終的に一括集約 nanoaggregation、形成するため従来の遊離基析出過程で観察されるものと一貫性のあるのとは対照的につながる種子ポリマーを増やすとを強調する必要があります。感温ナノゲル3。加熱時間の短縮はより小さいを形成しより多くの単分散粒子の良好な状況がまた発見されました。我々 は前駆体ポリマーの一方または両方が相対的にヒドラゾン結合の疎水性が増加ナノゲル衝突に集計の確率を高める LCST 上の温度で長い時間で、nanoaggregate を開催するその仮説します。いずれかの前駆体アルデヒドやヒドラジドする官能基この集約の架橋達成度が増加する可能性が高い。最終的には、加熱時間の短縮は、プロセスの観点から有利な架橋ポリマー添加; 後 2 分ほどで単分散ナノゲル人口を形作ることができます。10 分は、一貫して高架橋ナノゲルの生産もしながら単分散ナノゲルを作り出すことができる最長の時間をことが判明しました。興味深いことに、メソッドは、ほぼ同じ粒径と粒径分布が異なる速度で混合またはも大量にプロセスのスケーリングから生じる混合に非常に敏感ではありません。当初、この結果で驚いている間そう話すナノゲルの生産を調整する熱力学の主要な役割。
低 polydispersities を達成するためにコロイドの安定性と、nanoaggregate の水和の程度は、キーの変数のように見えます。たとえばより少なく親水性アルデヒド官能基化ポリマーではなく種として親水性酸ヒドラジド機能性高分子を用いたハイパーラマンは有意に低い polydispersities とナノゲルに します。実験アセンブリ温度と種子高分子の LCST の差も重要です。シード高分子 LCST 直上の温度で動作 ((T-LCST) < 5 ° C) 単分散ナノゲル形成の最も高い確率を提供していますLCST 結果を効果的にすることができない比較的コンパクトな種子ポリマーで以下の動作中に集計し、架橋するほど可能性が高くより多くの疎水性と折りたたまれたハイパーラマンを作成、LCST を上回る営業または再現性をもって架橋。状単分散粒子の最良の予測の最初核ポリマーの LCST 発症を測定する紫外/可視スキャンを実行してその後 1-2 ° C の温度で自己組織化プロセスを実行することをお勧めその LCST 上。
ナノゲルのこのメソッドを使用して生成の凍結乾燥し、コロイド安定性、自己組織化構造や架橋安定化手法に起因する我々 のビューでしばしば不可能に変更せず粉できることに注意してください。また種子ポリマーのみが動作するこのメソッドの感温する必要があると見込んでください。非対応または他の刺激への応答は、ポリマーを架橋の使用はさらにこの方法の究極の適用を広げるかもしれない。最後に、2 つの反応前駆体ポリマーの混合ですのでこの場合とは対照的なゲル化時間パッシブは説明他の製造方法に対するプロセス制御の面で大いにより少なく重要。ただし、この手法でさえ総架橋時間を維持する < 30 分は粒子凝集の危険性を最小限にすることが望ましい。
反応性エレクトロスピニング仁科ヒドロゲル
反応性の前の高分子のゲル化時間の制御は再びゲル ナノファイバー製造の成功に不可欠であります。特に、約一括ゲル化と (ダブルバレル シリンジと同様に長さとスタティック ミキサーの蛇行からソリューションの流量を変更することによって制御される) スタティック ミキサーは、前駆体ポリマーの滞留時間のマッチング前駆体ポリマーの時は針とコレクター間の紡糸繊維の効果的な架橋を確保できるよう曳糸性の維持の両方に不可欠です。高速ゲル化効果がテイラーの円錐形の開発につながる、従って遅いゲル化結果が広がり、その結果コレクターを打つゲルの代わりに水溶液で中の貧しい曳糸性と薄膜形成の究極の代わりにゲルナノファイバー。一括ゲル化時間滞留時間弱で働いてが有効 (および確かに針の目詰まりのリスクを軽減することが望ましい) をまた発見されています水の蒸発ソリューションは、効果的に回されるように集中して前駆体高分子のため、ストリームし、回転処理中にゲル化速度を加速します。高針・ コレクターの距離でこれと同じ調子で (> 10 cm) はこのプロセスで一般的に良好な短い距離水の蒸発時間を削減し、したがって関係でより厳格なコントロールを必要と滞留時間とゲル化時間ナノ製品を維持するために。
注意してください PEO の使用 (または別高分子量かつ容易にエレクトロスパンナノファイバー高分子) 短くて高い分岐 POEGMA オリゴマーを誘発するエンタングルメントの適切な学位を達することができない単独では、ナノファイバーの形成を促進するためにこのプロトコルで必要であります。エレクトロスピニング;代わりに、エレクトロ スプレーの結果はすべてで (ただし、これは、この同じ化学を用いた分解性ゲル粒子を作るためのアプリケーションもあります) POEGMA 専用製剤のテスト条件を処理します。1 wt % (1 MDa 分子量) の最小 PEO 濃度が完全にナノ形態を維持するために必要です。仁科ネットワークの整合性を中断させることがなく次の簡単な浸漬手順 (脱イオン水, 24 h) 繊維から、PEO を削除できることに注意してください。このように、PEO は最終的なナノ製品の不可欠なコンポーネントよりも過渡エレクトロスピニング援助としてより機能します。この同じと組み合わせて、さまざまな種類 (浸漬時にコレクターから剥離できる薄層ゲルを作成する) にシンプルなアルミ箔、アルミ円板 (厚い足場を作成) するなど、コレクターのことにも注意してください。法、ゲル化の速度、エレクトロスピニング法, 率とエレクトロスピニングの中に水の蒸発速度を制御する他のプロセス変数のまま不変です。
興味深いことに、異なる形態を準備するために使用する方法、に応じて同じヒドロゲル前駆体から作製したハイドロゲルの劣化時に有意な差を観測されています。たとえば、POEGMA ナノ ヒドロゲルは有意に高い表面積およびこうしてヒドラゾン結合を加水分解する水へのアクセスにもかかわらず同じ組成のバルク POEGMA ゲルより遅い低下します。我々 の関係間混合前駆体ポリマー内部のゲルの homogeneities につながる可能性がありますやが大幅に異なる形態やでの幾何学の観点から記述されていたプロトコル固有の対照のこれらの違いその場ゲル化、同時の水の蒸発とこのプロセスにおいて架橋によるエレクトロスピニングに特に関連として同じ時間スケールでポリマー前駆体の濃度。一方、各プロトコルで使用するターゲットが 1 つのポリマーの場合、前駆体ポリマーの選択これにやや複雑になる、それも 1 つの化学組成が非常に異なった物理的性質のゲルを作るという点で技術的な機会を提供するかもしれない。
全体的にみて、説明方法は感温性高分子 (一括、マイクロ、ナノ) 複数の長さスケールと複数の種類の内部構造の分解 (または少なくとも renally クリア可能な) 類縁体を製造するための戦略を提供する (粒子または繊維)。このようなプロトコル アドレス バイオメディカル分野に従来準備合成感温材料の巧妙な翻訳に重要な障壁: injectability と分解。薬剤配達および組織エンジニア リング アプリケーションの癌、物理ターゲットに至る血液脳関門、蛋白質の治療薬デリバリー薬物の輸送の両方でそのような材料の応用の探索を進めています目、その他のアプリケーションの間で、細胞の分化と組織の一方向成長可逆接着の背面。
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
自然科学工学研究会議のカナダ (レベル)、レベルの作成から資金調達-同上 (細胞外マトリックスの統合デザイン) プログラム 20/20: レベル眼生体材料研究ネットワーク、およびオンタリオ研究省と技術革新の初期研究者賞プログラムが認められています。
Materials
| Chemicals | |||
| 2,2 – azobisisobutryic acid dimethyl ester | Wako Chemicals | 101138 | |
| Di(ethylene glycol) methyl ether methacrylate (M(EO)2MA) | Sigma Aldrich | 447927 | 188.2 g/mol, n=2 ethylene oxide repeat units |
| Oligo (ethylene glycol) methyl ether methacrylate (OEGMA475) | Sigma Aldrich | 447943 | 475 g/mol, n=8-9 ethylene oxide repeat units |
| Acrylic acid (AA), 99% | Sigma Aldrich | 147230 | |
| Thioglycolic acid (TGA), 98% | Sigma Aldrich | T3758 | |
| Dioxane, 99% | Caledon Labs | 360481 | |
| Nitrogen, UHP grade | Air Liquide | Alphagaz1 765A-44 | |
| Adipic acid dihydrazide (ADH), 98% | Alfa Aesar | A15119 | |
| N'-ethyl-N-(3- dimethylaminopropyl)-carbodiimide (EDC, x%) | Carbosynth | FD05800 | |
| Hydrochloric acid (HCl), 37% | Sigma Aldrich | 320331 | |
| Sodium hydroxide (NaOH), 97% | Sigma Aldrich | 221465 | |
| Aminoacetyl aldehyde dimethyl acetal, 99% | Sigma Aldrich | 121967 | |
| 4-Hydroxy-TEMPO, 97% | Sigma Aldrich | 176141 | |
| Methacryloyl chloride,97x% | Sigma Aldrich | 523216 | |
| Petroleum ether, 95% | Sigma Aldrich | 32047 | |
| Magnesium sulfate, 99.5% | Sigma Aldrich | M7506 | |
| tert-Butyl methyl ether, >99.0% | Sigma Aldrich | 443808 | |
| Phosphate buffered saline | BioShop | PBS405.1 | 1x, pH 7.3-7.5 |
| N-isopropylacrylamide, 99% | J&K Scientific | 258717 | Recrystallized from 60% hexanes/40% toluene |
| Ethanol, anhydrous | Commerical Alchols | P016EAAN | |
| Span 80 | Sigma Aldrich | S6760 | |
| Heavy paraffin oil | Caledon Labs | 1326197 | |
| Pentane, reagent grade | Caledon Labs | 1/10/7800 | |
| Poly (ethylene oxide) average Mv 600,000 | Sigma Aldrich | 182028 | |
| Supplies essential for synthesis and hydrogel fabrication | |||
| Rotary evaporator | Heidolph | G3 | |
| Dialysis tubing (3500 Da molecular weight cut-off) | Spectrum Labs | 28170-166 | Vol/length= 6.4mL/cm |
| Double barrel syringe | Medmix | L series | L series, 2.5 mL, 1:1 volume ratio |
| Static mixer | Medmix | L series | L series, 2.5 mL, 1:1 volume ratio, 1.5" length |
| Silicone rubber sheet, 1/16" thickness | McMaster-Carr | 9010K12, 30A Durometer (Super Soft) | |
| Syringe pump | KD Scientific | KDS Legato 200 | Infuse Only Dual Syringe Pump |
| High voltage power supply | Spellman | 230-20R | 0 to 20 kV |
| Microfluidic Chip Fabrication | |||
| Silicon wafer | University Wafer | 2080 | D = 76.2 mm; 380 µm thickness; P-doped; <100> orientation |
| SU-8 100 | MicroChem | Y131273 | |
| SU-8 Developer | MicroChem | Y020100 | |
| Custom 2.5" spincoater | Built in-house | N/A | |
| Mask Aligner | KARL SUSS | MJB3 UV400 (with a 276 W lamp) | |
| Masterflex L/S 13 Silicone Tubing | Cole Parmer | OF-96400-13 | Peroxide-cured |
| Dow Corning Sygard 184 Silicone Elastomer Base | Ellsworth Adhesives | 4019862 | |
| Dow Corning Sygard 184 Silicone Elastomer Curing Agent | Ellsworth Adhesives | 4019862 | |
| High Power Plasma Cleaner | Harrick | PDC-002-HP | |
| Characterization Instruments | |||
| Mach 1 micromechanical tester | Biomomentum | LB007-EN | |
| Cellstar tissue culture 12 well plate | Greiner Bio-one | 665 180 | |
| Cell culture insert for 12 well plate | Corning | 08-771-12 | 8 µm pore size |
| Optical microscope | Olympus BX51 optical microscope | BX51 | |
| Temperature-controlled microscope stage | Linkam Scientific | THMS600 | |
| Gel permeation chromatograph (GPC) | Waters | 590 HPLC Pump | Waters Styragel columns (HR2, HR3, HR4; 30 cm x 7.8 mm (ID); 5 mm particles), Waters 410 refractive index detector |
| Dynamic light scattering (DLS) | Brookhaven | 90Plus Particle Size Analyzer | |
| Transmission electron microscopy (TEM) | TEMSCAN | JEOL 1200EX | Accelerating voltage 100 kV |
| Scanning electron microscopy (SEM) | Tescan | Vega II LSU | Accelerating voltage 10 kV |
| Microsquisher | CellScale Biomaterials Testing | MS-50M-01 |
Riferimenti
- Heskins, M., Guillet, J. E. Solution Properties of Poly(N-isopropylacrylamide). J. Macromol. Sci. A. 2 (8), 1441-1455 (1968).
- Lutz, J. -. F., Akdemir, &. #. 2. 1. 4. ;., Hoth, A. Point by Point Comparison of Two Thermosensitive Polymers Exhibiting a Similar LCST: Is the Age of Poly(NIPAM) Over. J. Am. Chem. Soc. 128 (40), 13046-13047 (2006).
- Pelton, R. H., Chibante, P. Preparation of Aqueous Lattices with N-Isopropylacrylamide. Colloids Surf. 20 (3), 247-256 (1986).
- Palasis, M., Gehrke, S. H. Permeability of Responsive Poly(N-Isopropylacrylamide) Gel to Solutes. J. Controlled Release. 18 (1), 1-11 (1992).
- Kawaguchi, H., Fujimoto, K., Mizuhara, Y. Hydrogel Microspheres .3. Temperature-Dependent Adsorption of Proteins on Poly-N-Isopropylacrylamide Hydrogel Microspheres. Colloid Polym. Sci. 270 (1), 53-57 (1992).
- Okuyama, Y., Yoshida, R., Sakai, K., Okano, T., Sakurai, Y. Swelling Controlled Zero-Order and Sigmoidal Drug-Release from Thermoresponsive Poly(N-Isopropylacrylamide-Co-Butyl Methacrylate) Hydrogel. J. Biomater. Sci. Polym. Ed. 4 (5), 545-556 (1993).
- Snowden, M. J. The Use of Poly(N-Isopropylacrylamide) Lattices as Novel Release Systems. J. Chem. Soc. – Chem. Comm. (11), 803-804 (1992).
- Haraguchi, K., Takehisa, T., Ebato, M. Control of cell cultivation and cell sheet detachment on the surface of polymer/clay nanocomposite hydrogels. Biomacromolecules. 7 (11), 3267-3275 (2006).
- Lee, B., et al. Initiated chemical vapor deposition of thermoresponsive poly(N-vinylcaprolactam) thin films for cell sheet engineering. Acta Biomater. 9 (8), 7691-7698 (2013).
- Cole, M. A., Voelcker, N. H., Thissen, H., Griesser, H. J. Stimuli-responsive interfaces and systems for the control of protein-surface and cell-surface interactions. Biomaterials. 30 (9), 1827-1850 (2009).
- Feil, H., Bae, Y. H., Feijen, J., Kim, S. W. Molecular Separation by Thermosensitive Hydrogel Membranes. J. Membrane Sci. 64 (3), 283-294 (1991).
- Kim, J., Park, K. Smart hydrogels for bioseparation. Bioseparation. 7 (4-5), 177-184 (1998).
- Yamashita, K., Nishimura, T., Nango, M. Preparation of IPN-type stimuli responsive heavy-metal-ion adsorbent gel. Polym. Adv. Tech. 14 (3-5), 189-194 (2003).
- Ziolkowski, B., Czugala, M., Diamond, D. Integrating stimulus responsive materials and microfluidics: The key to next-generation chemical sensors. J. Intelligent Mater. Syst. Struct. 24 (18), 2221-2238 (2013).
- Zhang, Y., Kato, S., Anazawa, T. A flap-type hydrogel actuator with fast responses to temperature. Smart Mater. Struct. 16 (6), 2175-2182 (2007).
- Suzuki, D., Taniguchi, H., Yoshida, R. Autonomously Oscillating Viscosity in Microgel Dispersions. J. Am. Chem. Soc. 131 (34), 12058-12059 (2009).
- Schild, H. G. Poly(N-isopropylacrylamide): Experiment, Theory and Application. Prog. Polym. Sci. 17, 163-249 (1992).
- Oh, J. K., Min, K., Matyjaszewski, K. Preparation of poly (oligo (ethylene glycol) monomethyl ether methacrylate) by homogeneous aqueous AGET ATRP. Macromolecules. 39 (9), 3161-3167 (2006).
- Vihola, H., Laukkanen, A., Tenhu, H., Hirvonen, J. Drug Release Characteristics of Physically Cross-Linked Thermosensitive Poly(N-vinylcaprolactam) Hydrogel Particles. J. Pharm. Sci. 97 (11), 4783-4793 (2008).
- Zhang, L. F., Liang, Y., Meng, L. Z. Thermo-sensitive amphiphilic poly(N-vinylcaprolactam) copolymers: synthesis and solution properties. Polym. Adv. Tech. 21 (10), 720-725 (2010).
- Smeets, N. M. B., Bakaic, E., Patenaude, M., Hoare, T. Injectable and tunable poly(ethylene glycol) analogue hydrogels based on poly(oligoethylene glycol methacrylate). Chem. Comm. 50 (25), 3306-3309 (2014).
- Lutz, J. -. F. Polymerization of oligo (ethylene glycol)(meth) acrylates: toward new generations of smart biocompatible materials. J. Polym. Sci. A. 46 (11), 3459-3470 (2008).
- Lutz, J. -. F., Hoth, A. Preparation of Ideal PEG Analogues with a Tunable Thermosensitivity by Controlled Radical Copolymerization of 2-(2-Methoxyethoxy)ethyl Methacrylate and Oligo(ethylene glycol) Methacrylate. Macromolecules. 39 (2), 893-896 (2006).
- Patenaude, M., Campbell, S., Kinio, D., Hoare, T. Tuning Gelation Time and Morphology of Injectable Hydrogels Using Ketone-Hydrazide Cross-Linking. Biomacromolecules. 15 (3), 781-790 (2014).
- Patenaude, M., Hoare, T. Injectable, Degradable Thermoresponsive Poly(N-isopropylacrylamide) Hydrogels. ACS Macro Lett. 1 (3), 409-413 (2012).
- Patenaude, M., Hoare, T. Injectable, Mixed Natural-Synthetic Polymer Hydrogels with Modular Properties. Biomacromolecules. 13 (2), 369-378 (2012).
- Smeets, N. M. B., Bakaic, E., Patenaude, M., Hoare, T. Injectable poly(oligoethylene glycol methacrylate)-based hydrogels with tunable phase transition behaviours: Physicochemical and biological responses. Acta Biomater. 10 (10), 4143-4155 (2014).
- Sivakumaran, D., Mueller, E., Hoare, T. Temperature-Induced Assembly of Monodisperse, Covalently Cross-Linked, and Degradable Poly(N-isopropylacrylamide) Microgels Based on Oligomeric Precursors. Langmuir. 31, 5767-5778 (2015).
- Bakaic, E., Smeets, N. M. B., Dorrington, H., Hoare, T. “Off-the-shelf” thermoresponsive hydrogel design: tuning hydrogel properties by mixing precursor polymers with different lower-critical solution temperatures. RSC Adv. 5 (42), 33364-33376 (2015).
- Bulpitt, P., Aeschlimann, D. New strategy for chemical modification of hyaluronic acid: Preparation of functionalized derivatives and their use in the formation of novel biocompatible hydrogels. J. Biomed. Mater. Res. 47 (2), 152-169 (1999).
- Kesselman, L. R. B., Shinwary, S., Selvaganapathy, P. R., Hoare, T. Synthesis of Monodisperse, Covalently Cross-Linked, Degradable “Smart” Microgels Using Microfluidics. Small. 8 (7), 1092-1098 (2012).
- Sivakumaran, D., Mueller, E., Hoare, T. Microfluidic production of degradable thermoresponsive microgels based on poly(N-isopropylacrylamide). Soft Matter. , (2016).
- Xu, F., Sheardown, H., Hoare, T. Reactive Electrospinning of Degradable Poly(oligoethylene glycol methacrylate)-Based Nanofibrous Hydrogel Networks. Chem. Comm. 52 (7), 1451-1454 (2016).
- Troll, K., et al. The collapse transition of poly(styrene-b-(N-isopropyl acrylamide)) diblock copolymers in aqueous solution and in thin films. Colloid Polym. Sci. 286 (8), 1079-1092 (2008).
- Patenaude, M., Campbell, S., Kinio, D., Hoare, T. Tuning Gelation Time and Morphology of Injectable Hydrogels Using Ketone-Hydrazide Cross-Linking. Biomacromolecules. 15 (3), 781-790 (2014).
- Kelly, T. A., Felder, M. S., Ollar, R. A. Inducing Apoptosis in a Mammalian Cell by Contacting with Paraffin or Agar. US Patent. , (2001).
