Fabrikasjon nedbrytbar Thermoresponsive Hydrogels på flere lengde skalaer via reaktive ekstrudering, Microfluidics, selvstendig montering, og Electrospinning
Summary
Protokoller er beskrevet for fabrikasjon av nedbrytbart thermoresponsive hydrogels basert på hydrazone cross-linking av polymere oligomers på bulk skalaen, Mikroskala, og nanoskala, sistnevnte for utarbeidelse av både gel nanopartikler og nanofibers.
Abstract
Mens ulike smarte materialer har blitt utforsket for en rekke biomedisinsk programmer (f.eks, narkotika-leveranser, vev engineering, bioimaging, osv.), vært deres ultimate klinisk bruk hemmet av mangel på biologisk relevante degradering observert mest smart materialer. Dette gjelder særlig for temperatur svarer hydrogels, som er nesten jevnt basert på polymerer som er funksjonelt ikke-nedbrytbare (f.ekspoly(N-isopropylacrylamide) (PNIPAM) eller poly (oligoethylene glycol methacrylate) (POEGMA) ). Slik effektivt oversette potensialet i thermoresponsive hydrogels til utfordringene med fjernstyrte eller metabolisme regulert narkotika-leveranser, celle stillaser med tunable celle-materiell samhandling, theranostic materialer med potensial for både bildebehandling og narkotika-leveranser og andre slike programmer er en metode nødvendig for å gjengi hydrogels (hvis ikke fullt nedbrytbart) minst i stand til nyresvikt klaring etter nødvendige levetiden av materialet. Derfor, beskriver denne protokollen utarbeidelsen av hydrolytically-nedbrytbar hydrazone-krysskoblet hydrogels på flere lengde skalaer basert på reaksjonen mellom hydrazide og aldehyd-functionalized PNIPAM eller POEGMA oligomers med molekylær vekter under den nyre filtrering grensen. Spesielt metoder å dikte nedbrytbar thermoresponsive bulk hydrogels (med en dobbel fat sprøyte teknikk), hydrogel partikler (på begge Mikroskala ved hjelp av en microfluidics plattform tilrettelegge samtidige miksing og emulgering forløper polymerer og nanoskala ved hjelp av en termisk drevet selvstendig montering og cross-linking metoden), og hydrogel nanofibers (med en reaktiv electrospinning strategi) er beskrevet. I hvert tilfelle, hydrogels med temperatur-responsive egenskaper lik de oppnådde via konvensjonelle frie radikaler cross-linking prosesser kan oppnås, men hydrazone krysskoblet nettverket kan svekkes over tid til nytt på oligomeric forløperen polymerer og Aktiver klarering. Slik forventer vi disse metoder (som kan være generelt gjelder alle syntetiske vannløselige polymer, ikke bare smart materialer) gjør det lettere oversettelse av smart kunsstoff klinisk programmer.
Introduction
Smarte materialer har tiltrukket betydelig oppmerksomhet på grunn av deres potensial for reversibel “on-demand” svar til eksterne og/eller miljømessige signaler. Temperatur-responsive materialer har tiltrukket interesse på grunn av sin lavere kritisk løsning temperatur (LCST) atferd, som resulterer i temperatur-drevet nedbør ved temperaturer T > LCST1,2. I forbindelse med thermoresponsive hydrogels, dette for lavere kritisk løsning-temperatur er manifestert av reversibel hevelse/de-swelling hendelser som er resultater i temperatur-tunable bulk størrelser (større på T < LCST)3, pore størrelse (større på T < LCST)4og interfacial egenskaper (mer hydrofile på T < LCST)5. Slike overganger har vært mye brukt i narkotika-leveranser (for ekstern eller miljømessig triggerable narkotika release4,6,7), vev engineering og celle kultur (for thermoreversible celleadhesjon / delaminering8,9,10), separasjoner (for valgbar membran porosities og permeabilities eller termisk-resirkulerbare diagnostiske støtter11,12, 13), microfluidic behandler (for på-av ventiler regulerer flyten14,15), og reologiske modifikatorer (for temperatur-tunable viskositet16). De vanligste undersøkt thermoresponsive hydrogels er basert på poly(N-isopropylacrylamide) (PNIPAM)17, selv om betydelige (og økende) arbeidet er også utført på poly (oligoethylene glycol methacrylate) (POEGMA)2 ,18 og poly(vinylcaprolactam) (PVCL)19,20. POEGMA har fått spesiell siste interesse gitt sin etterlengtede forbedret biocompatibility21,22og virkemåten lettvinte-til-tune LCST i hvilke lineært forutsigbar blandinger av monomerer med ulikt antall etylen oksid gjenta enheter i sine siden kjeder kan endre LCST fra ~ 20 ° C til > 90 ° C2,23. Imidlertid hver av disse polymerer er utarbeidet av frie radikaler polymerisasjon og dermed inneholder en karbon-karbon ryggrad, betydelig begrense potensielle av og translatability av slike polymerer i forbindelse med biomedisinsk applikasjoner der nedbrytning (eller minst kapasiteten for fortolling gjennom nyre filtrering) er vanligvis et krav.
Svar på denne begrensningen, har vi nylig rapportert omfattende på anvendelse av hydrazone kjemi (dvs., reaksjonen mellom hydrazide og aldehyd-functionalized før polymerer) å forberede nedbrytbart analoger av thermoresponsive hydrogels24,25,26,27,28,29. Hydrazide og aldehyd grupper ved blanding av functionalized forløper polymerer30 rask og reversibel reaksjonen kan både i situ gelation (aktivere lettvinte injeksjon av dette materialet uten behov for kirurgiske implantering eller typer eksterne polymerisasjon stimulans som UV bestråling eller kjemiske innvielse) samt hydrolytisk nedbrytning av nettverket frekvensen kontrollert av kjemi og tetthet av crosslinking nettsteder. Videre ved å molekylvekt av at pre polymerene å forberede hydrogels under den nyre filtrering grensen, forringes hydrogels med denne tilnærmingen tilbake til de oligomeric forløper polymerer som kan fjernes fra kroppen25 ,27,28. Sammen med lav cytotoksisitet og lav inflammatorisk vev svar av materialer25,26,27, tilbyr denne tilnærmingen en potensielt oversettbare metode for bruk av thermoresponsive Smart hydrogels i medisin, spesielt hvis godt kontrollerte nedbrytbart analoger av slike hydrogels på alle lengden skalaer (bulk, mikro og nano) kan fremstille.
I denne protokollen beskriver vi metoder for å lage syntetiske thermoresponsive pre polymerer functionalized med kontrollert antall hydrazide og aldehyd grupper samt metoder å bruke disse polymerer opprette hydrogels med godt definerte dimensjonene på ulike lengde skalaer. Spesielt dette manuskriptet beskriver fire forskjellige metodene vi har utviklet for å styre en blanding av reaktive hydrazide og aldehyd-functionalized før polymerer og dermed skape thermoresponsive hydrogel nettverk med veldefinerte geometrier og morphologies:
For å lage nedbrytbar bulk hydrogels med definerte størrelser, en templating strategi er beskrevet som de reaktive pre polymerer lastes inn i separate fat dobbel fat sprøyte utstyrt på sitt utløp med en statisk mikser og deretter co ekstrudert i en silikon mold med de ønskede hydrogel form og dimensjoner21,27 (figur 1).
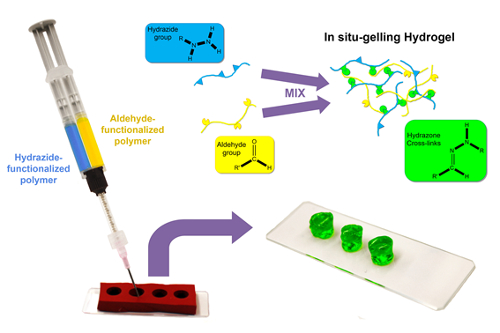
Figur 1 : Skjematisk bulk hydrogel formasjonen. Hydrazide og aldehyd-functionalized polymer løsninger (i vann eller vandig buffer) er lastet inn i separate fat dobbel fat sprøyte og deretter co ekstrudert gjennom en statisk mikser i sylindriske silikon mold. Rask i situ gelation på blande former en hydrazone krysskoblet hydrogel, som er frittstående (når mold er fjernet) innen sekunder til minutter avhengig av konsentrasjon og funksjonsgruppe tetthet av forløperen polymerer. Klikk her for å se en større versjon av dette tallet.
For å lage nedbrytbar gelépartikler på mikro-skala, reaktive microfluidics metode beskrives som forløper polymer løsninger er samtidig blandet og emulgert bruker en myk litografi mal microfluidic chip-design, slik at den dannelsen av blandet reaktive polymer dråper som senere gel i situ skjemaet gel microparticles med størrelser mal av emulsjon (figur 2)31,32.
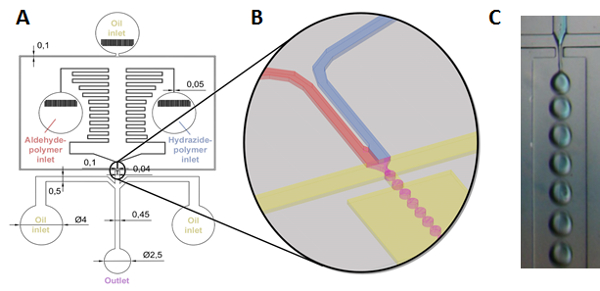
Figur 2 : Skjematisk av gel microparticle formasjon via reaktive microfluidics. (A, B) Hydrazide og aldehyd-functionalized polymer løsninger (i vann eller vandig buffer) er lei av sprøytepumpe i separate reservoarer som kobles nedstrøms over sikk-sakk flere kanaler designet for å skape en trykkgradient hindre tilbakestrømning. Polymerer deretter blandet like før blir skåret av parafinolje strømmer fra begge sider (også kjørt av en sprøytepumpe) og tvunget gjennom en dyse, resulterer i flyt-fokus produksjon av vandig (polymer løsning) dråper i en kontinuerlig parafin olje fase (se (B) for en illustrasjon av munnstykket og slippverktøy formasjon prosessen). En ytterligere to parafin olje innganger er plassert etter munnstykket til ytterligere separat dråper i samlingen kanalen å tillate komplett gelation før partikkel fjerning fra laminær strømning, hvoretter den resulterende microparticulate gels er samlet i et magnetisk rørt beaker; (C) bilde av slippverktøy prosessen ved munnstykket (Merk at hydrazide polymer er merket blå å illustrere blande)
Lage nedbrytbar gelépartikler på nanoskala, en termisk drevet reaktive selvtillit forsamlingen metoden er beskrevet som en løsning på en av reaktive forløper polymerer (“frø” polymer) varmes over sin LCST til en stabil nanoaggregate som er senere krysskoblet med tillegg av den komplementære reaktive forløper polymer (“crosslinking” polymer); den resulterende hydrazone krysskoblet nanogel har en størrelse mal direkte av nanoaggregate (Figur 3)28.
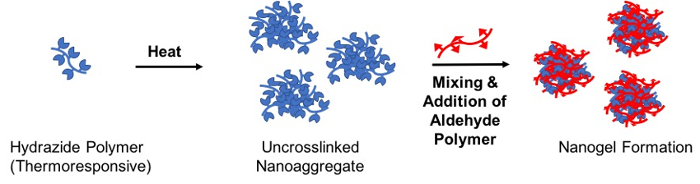
Figur 3 : Skjematisk av nanogel formasjon via termisk-drevet reaktive selvtillit forsamlingen. En vannløsning inneholdende (thermoresponsive) hydrazide-functionalized polymer varmes over sin lavere kritisk løsning temperatur å lage en stabil uncrosslinked nanoaggregate. Etter, en aldehyd-functionalized polymer legges til krysskobling nanoaggregate via hydrazone bond dannelse og dermed stabilisere nanogel partikkel på kjøling under LCST. Klikk her for å se en større versjon av dette tallet.
For å lage nedbrytbar nanofibers, er en reaktiv electrospinning teknikk beskrevet som en dobbel fat sprøyte utstyrt med en statisk mikser på sitt utløp (som brukes til å lage bulk hydrogels) er koblet til en standard electrospinning plattform (Figur 4 )33.
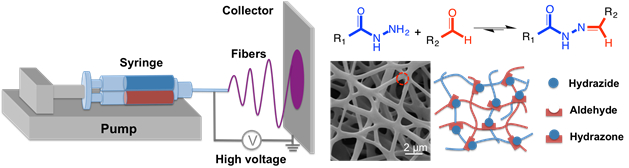
Figur 4 : Skjematisk av hydrogel nanofiber formasjon via reaktive electrospinning. En dobbel fat sprøyte med en statisk mikser (lastet som bulk hydrogels men også inkludert en brøkdel av høy molekylvekt poly(ethylene oxide) som et electrospinning Hjelpemiddel) er montert på en sprøytepumpe med nålen på slutten av koblet til en høy spenning strømforsyning. Hydrazone crosslinking oppstår under fiber spinning prosessen slik at når strømmen treff collector (aluminiumsfolie eller en roterende aluminium disk) nanofibrous morfologi opprettholdes. Klikk her for å se en større versjon av dette tallet.
Bruk av slike metoder for å lage nedbrytbar smart hydrogel nettverk demonstreres i denne protokollen bruker enten PNIPAM eller POEGMA som polymer steder. men de grunnleggende metodene beskrevet kan oversettes til en vannløselig polymer, men med passende justeringer for viskositet og (i tilfelle av den selv-montering nanogel fabrikasjon metoden) stabiliteten i den pre polymer i forming frø nanoaggregate.
Protocol
Representative Results
Discussion
Vi har brukt alle disse fabrikasjon teknikker for flere polymer systemer bruker bare små variasjoner av metodene som er beskrevet i detalj over for PNIPAM og POEGMA; brukere av disse protokollene må imidlertid være cognizant av den potensielle problemer som kan oppstå når andre polymerer erstattes i disse prosessene. Spesielt kan øke viskositeten av forløperen polymerer negativt påvirke både processibility (spesielt i metoden microfluidic) samt effektiviteten av blanding av to forløper polymerer. I tillegg må gelation da polymerer kontrolleres frekvensen avhengig av morfologi målrettet for å unngå tidlig gelation som tjener til å hemme flyt eller hindre interdiffusion av de reaktive pre polymerer, viktig til ønsket homogen gel strukturer. Spesifikke begrensninger av hver strategi, samt tilnærminger vi har brukt til å tilpasse disse tilnærminger til adressen slike begrensninger på hver fabrikasjon lengde skala, er beskrevet nedenfor.
Bulk hydrogels via dobbel fat sprøyte co-ekstrudering
Gelation er viktige variabelen til kontroll for å sikre effekten av dobbel fat sprøyte teknikken for å danne bulk hydrogels. Polymerer som gel for fort ved kontakt ( 5 s er å foretrekke (selv om ikke nødvendig) for bruk av denne teknikken; Dette er spesielt viktig hvis Repliker hydrogels støpes for fysisk eller mekanisk analyse å sikre at hver hydrogel cast har samme sammensetning. Gelation tid kan enkelt endres ved å endre tettheten av reaktive funksjonelle grupper på en eller begge forløper polymerer (lavere funksjonsgruppe tetthet fører til tregere gelation) eller endre konsentrasjonen av forløperen polymerer brukes til gel ( lavere konsentrasjoner fører til tregere gelation)21. Eventuelt reduserer erstatte (mer reaktiv) Aldehydiske med gruppen (mindre reaktiv) keton som electrophile i gelling par betydelig gelation uten betydelig endre sammensetningen av den resulterende hydrogel35 ; polymerer tilberedt med blandinger av aldehyd og keton monomerisk forløpere kan brukes til å justere gelation tiden som du ønsker uten å endre konsentrasjonen av forløperen polymerene (og dermed masse prosentandelen av faste stoffer i resulterende gel dannet).
Vi vil også oppmerksom på at første hydrogel kastet ikke alltid har de samme egenskapene som etterfølgende hydrogels kastet, en observasjon tilskrevet små forskjeller i prisen som innholdet i to fat faktisk nå statisk blandebatteri. Som et resultat vi vanligvis prime dobbel fat sprøyten ved ekstrudering en liten (< 0,3 mL) brøkdel av gel før starte støping prosessen for å minimere slike variasjon. Endelig, mens ikke vanligvis problematisk når du bruker oligomeric syntetiske pre polymerer, viskositeten av en eller flere forløper polymer løsninger kan utgjøre en utfordring i sammenheng med denne teknikken, både når det gjelder tilrettelegging flyt med enkel tommelfingeren nedgangstiden og fremme effektiv blanding i statisk blandebatteri. Men noe overraskende, selv forløper polymer løsninger med kraftig forskjellige viskositet utgjør fremdeles relativt homogene hydrogels bruker statisk blandebatteri vedlegg beskrives i listen deler (f.eks PNIPAM med en høy molekylær vekt karbohydrater26), foreslå det gjelder ineffektiv blande som følge av mis matchet viskositet kan ikke være betydelig minst på bulk skala. Hvis nødvendig, kan bruk av en sprøytepumpe (i stedet for tommelen) til stasjonen flyt og/eller bruk av en større gauge nål ved uttaket hjelpe overvinne problemer forbundet med extrudability i disse systemene.
Mikroskala hydrogels via reaktive microfluidics
Det viktigste trinnet forbundet med microfluidics tilnærming gel microparticle fabrikasjon er fylling av microfluidics chip med de to reaktive polymerer. Hvis polymerer leveres med ulike trykk eller med forskjellige satser til chip, differansetrykket kan kjøre tilbakestrømming av en forløper polymer løsning i reservoaret (eller minst mot reservoaret) av den andre forløper polymer. Dette resulterer i gelation oppstrøms fra partikkel formasjon, effektivt blokkerer flyten, og dermed krever chip disposisjon. Forferdelige banen trykt mellom hver reservoaret og miksing punktet skaper en betydelig motstand mot tilbakestrømming; men vil selv en utdannet operatør noen ganger gel en chip før en stabil regime er oppnådd. Basert på vår erfaring, er mellom 1-2 min vanligvis nødvendig for å stabilisere renn følgende initiering av slippverktøy dannelse (over da relativt polydisperse gel microparticles produseres); Hvis ingen problemer overholdes i de første 5-10 minuttene av drift, er det sannsynlig at flere timer med kontinuerlig monodisperse partikkel produksjon kan oppnås. Bruk av forløperen polymerer med relativt godt matchet viskositet samt ikke-øyeblikkelig gelation ganger (minst > 15 s å foretrekke) sterkt bistår unngå slike problemer og fremme dannelsen av stabil renn.
Merk at ulike flow priser fra 0.01-0,1 mL/h i den vandige fasen og 1.1-5.5 mL/t i olje fasen har blitt testet med denne chip-design, fører til fabrikasjon av partikler på størrelse rekke ~ 25-100 µm ifølge skråstillingen på den flyt-fokus veikryss; raskere flyt priser likestille til høyere skjær og dermed mindre partikler dannet31,32. Varierende olje infusjonshastigheten samtidig totale vandig flow rate lav (~0.03 mL/t, som sitert i protokollen) ble funnet for å være mest effektivt å kontrollere gel microparticle størrelse uten å kompromittere enten monodispersity eller levetiden til enheten, var begge observert å betydelig redusere på høyere enden av de siterte totale vandig strømningshastigheter. Større olje flyt priser (> 5,5 mL/t) opprette mindre partiklene er mulig, men økte risikoen for chip delaminering (en vanlig begrensning møtte med plasma-limt PDMS microfluidic sjetonger). Liming sjetongene en annen metode kan aktivere raskere flyt priser og dermed mindre gel microparticle produksjon, en strategi som vi er for tiden utforske. Redusere munnstykket kan også bidra til å redusere størrelsen på microparticles som kan produseres, om enn på en økt risiko for tidlig gelation før partikkel formasjon. Tregere strømningshastigheter tendens til å føre til flyt ustabilitet og dermed høyere polydispersities og økt risiko for chip gelation; Denne begrensningen kan overvinnes ved hjelp av en flerkanals microfluidic flyt control system som har høyere stabilitet og høyere oppløsning enn standard sprøyte pumpene som brukes i denne protokollen.
Valget av olje var avgjørende for suksessen til denne protokollen, som tyngre olje (gunstig i forebygging gel microparticle agglomeration etter samling) førte til mye mindre konsekvente partikkel dannelsen ved munnstykket enn lys silikonolje rapportert i protokollen. Vi hypothesize dette redusert reproduserbarhet er et resultat av lavere konsistensen av sprøyte pumping av tyngre olje, fører til mer variabel skjær på blande punktet. Unngå gel microparticle samling i samling flasken ble også en utfordring, spesielt umiddelbart ved utkjørselen fra microfluidic enheten da i situ gelation ikke var fullstendig og store antall tilgjengelige reaktive funksjonelle grupper var tilgjengelig for skjemaet broer mellom kolliderer partikler i samlingen badekaret. Denne utfordringen er rettet: øke lengden på exit kanal på microfluidic chip selv, opprettholde gel microparticles i laminær strømning for en lengre periode å fremme mer komplett gelation; legger til side kanalene etter munnstykket å mate mer olje i chip og dermed bedre separat gel microparticles i denne post blande kanalen uten å påvirke den skjær felt på munnstykket selv eller den partikkel produksjon; og legge en magnetisk mikser til samling kolbe å unngå gel microparticle sedimentering og vedlikeholde et større gjennomsnittlig skille mellom tilstøtende partikler. Mens svært langsom gelling-polymerer ville sannsynligvis forbedre enheten stabiliteten og minimere problemer med grunning, ble slike systemer også observert å betydelig øke risikoen for gel microparticle aggregering, som et større antall reaktive funksjonelle grupper forblir Ureagert (og dermed kunne form mellom partikkel broer) over en lengre periode. Slik gelation ganger på 15-60 s synes å være optimal for denne teknikken: sakte nok til å aktivere grunning men rask nok å sikre mest reaktive funksjonsgrupper forbrukes før gel microparticles avslutter laminær strømning kanalen i det samling kolbe.
Endelig er fjerning av templating oljen viktig å sikre at den resulterende partikler opprettholde egenskapene smart forventet basert på sammensetningen av de pre polymerer lagt og aktiverer bruk av disse partiklene i biomedisinsk sammenheng. Pentane vask fremgangsmåten var effektive i denne forbindelse for generelle gel microparticle produksjon. Bruk av denne teknikken i en direkte biomedisinsk sammenheng (f.eks, på prosessoren celle innkapsling) krever imidlertid revurdering av denne protokollen. Vi har også utforsket bruk av olivenolje, foreslått for å være en mer inert olje i sammenheng med å kontakte celler36, som i dispergerings. Mens partikkel formasjon var mulig, var gel microparticle befolkningen betydelig mer polydisperse enn kan oppnås med mineralolje, minst med gjeldende chip design. Således, mens chip synes å være tilpasningsdyktige både Syntetisk polymer og naturlig polymer gel microparticle formasjon31, en modifisert design kan være nødvendig å utnytte denne teknikken videre over alle mulige materiale kombinasjoner.
Nanoskala hydrogels via reaktive selvstendig montering
Nanogels er dannet ved hjelp av et svært bredt spekter av behandlingen forhold, herunder ulike konsentrasjoner av frø polymer (0,5-2 wt %), ulike forhold av crosslinking:seed polymer (0,05-0.2), forskjellige temperaturer (40-80 ° C), ulike blande hastigheter ( 200-800 rpm), og ulike oppvarming ganger etter tillegg av crosslinker polymer (2-60 minutter)28. I konsentrasjoner er observerte trender generelt som ville bli spådd, høyere konsentrasjoner av frø polymer føre til større nanogels og høyere prosenter av crosslinker:seed polymer føre til nanogels med høyere krysskobling tettheter og dermed lavere thermoresponsivities. Det bør understrekes at økende seedet polymer konsentrasjon for høy til slutt fører til bulk aggregering i motsetning til nanoaggregation, konsistent med hva er observert i konvensjonelle frie radikaler nedbør prosessen for å danne thermoresponsive nanogels3. Kortere oppvarming ganger ble også funnet for å være gunstig for forming mindre og mer monodisperse partikler. Vi hypothesize at holder nanoaggregate på lengre tid ved en temperatur over LCST ett eller begge av forløperen polymerer øker sannsynligheten for samling på nanogel kollisjon, med økt hydrophobicity av hydrazone obligasjonen forhold til enten de forløper aldehyd eller hydrazide funksjonelle gruppene gjør denne aggregering mer sannsynlig som graden av crosslinking oppnådd er økt. Til slutt, kortere oppvarming ganger er gunstige fra en prosess perspektiv, som monodisperse nanogel innbyggere kan dannes i så lite som 2 minutter etter crosslinker polymer; 10 min ble funnet for å være den lengste tiden som kunne produsere monodisperse nanogels samtidig for produksjon av flere svært krysskoblet nanogels. Interessant, er metoden bemerkelsesverdig ufølsom for miksing, med nesten identiske partikkelstørrelser og partikkel størrelse distribusjoner som følge av miksing på ulike hastigheter selv skalering prosessen til større volumer. Mens opprinnelig overrasket med dette resultatet, taler det sannsynlig til hovedrollen Termodynamikkens i å regulere nanogel produksjon.
For å oppnå lav polydispersities, synes kolloidal stabiliteten og graden av hydrering av nanoaggregate å være viktige variabler. For eksempel føre nanoaggregates tilberedt med de mer hydrofile hydrazide-functionalized polymerer som frø i motsetning til de mindre hydrofile aldehyd-functionalized polymerer til nanogels med betydelig lavere polydispersities. Forskjellen mellom eksperimentelle montering temperaturen og LCST av frø polymer er også kritisk. Ved en temperatur like over frø polymer LCST ((T-LCST) < 5 ° C) tilbyr den høyeste sannsynligheten av monodisperse nanogel formasjon; fungerer godt over LCST skaper mer hydrofobe og skjult nanoaggregates som er mer sannsynlig til samlet og mindre sannsynlig å krysskobling, mens opererer under LCST resultatene i en relativt ikke-kompakt frø polymer som ikke kan effektivt eller reproduserbar krysskoblet. For beste prediksjon av partikkel monodispersity, anbefaler vi først utføre en UV/vis skanning for å måle utbruddet LCST av frø polymer og senere utføre selvtillit forsamlingen behandle en temperatur 1-2 ° C over at LCST.
Merk at nanogels produsert ved hjelp av denne metoden kan være lyofiliserte og redispersed uten endring i kolloidalt stabilitet, ofte ikke mulig for selv montert strukturer og tilskrives for vår crosslinking stabilisering metode. Vi forventer at bare den frø polymer må være thermoresponsive for denne metoden skal fungere; Bruk av cross-linking polymerer som er utilgjengelig eller lydhør overfor andre stimuli kan ytterligere utvide den ultimate anvendelsen av denne teknikken. Til slutt, siden blanding av de to reaktive forløper polymerer er i dette tilfellet passiv i motsetning til aktiv, gelation tid er mye mindre viktig i prosessen kontrollen i forhold til de andre fabrikasjon strategiene beskrevet. Men selv i denne teknikken, holder den totale crosslinking tiden < 30 min er ønskelig å minimere risikoen for partikkel aggregering.
Nanofibrous hydrogels via reaktive electrospinning
Kontrollere gelation da de reaktive pre polymerer er igjen avgjørende for suksessen av gel nanofiber produksjon. Spesielt ca matchende oppholdstiden forløper polymerer i statisk mikseren (kontrollert av endre infusjonshastigheten løsning fra dobbel fat sprøyten som lengden og tortuosity av statisk blandebatteri) med bulk gelation forløperen polymerer er viktig både for å bevare spinnability, samt sikre effektiv crosslinking av spunnet fibrene mellom nålen og samler. Raskere gelation fører til ineffektiv Taylor kjegle utvikling og dermed dårlig spinnability, mens tregere gelation resultater i en vandig løsning i stedet for en gel treffer collector, noe som resulterer i spredning og den ultimate dannelsen av en tynn film gel i stedet for nanofibers. Arbeider på residence ganger litt nedenfor bulk gelation tiden har også funnet for å være effektiv (og faktisk foretrekke å redusere risiko for p clogging) siden vannet fordamper som løsningen er spunnet effektivt konsentrerer forløper polymerer i den streame og dermed akselererer gelation kinetics under spinning prosessen. I denne samme ånd, opererer på høyere p-til-samler distanser (> 10 cm) er generelt gunstige i denne prosessen, kortere distanser redusere tiden tilgjengelig for fordampning og dermed kreve strengere kontroll over forholdet mellom botid og gelation tid for å bevare et nanofibrous produkt.
Merk at bruk av PEO (eller annen molekylvekt og enkelt electrospun polymer) er avgjørende i denne protokollen å fremme nanofiber formasjon, som kort og svært forgrenet POEGMA oligomers ikke alene når en tilstrekkelig grad av sammenfiltring å indusere electrospinning; i stedet behandle electrospray resultater på alle forhold testet for POEGMA bare formuleringer (selv om dette har også programmer for å lage nedbrytbar gelépartikler bruker denne samme kjemi). En minimum PEO konsentrasjon av 1 wt % (1 MDa molekylvekt) er nødvendig for å opprettholde en fullt nanofibrous morfologi. Merk at PEO kan fjernes fra papirfibrene etter en enkel soaking prosedyre (deionisert vann, 24 h) uten å forstyrre integriteten til nanofibrous nettverket; på denne måten fungerer PEO mer som en forbigående electrospinning hjelp enn en vesentlig komponent i siste nanofibrous produktet. Merk også at ulike samlere, inkludert enkel aluminiumsfolie (for å lage tynt lag hydrogels som kan delaminate fra collector på soaking) samt en roterende aluminium disk (for å opprette tykkere stillaser) kan brukes sammen med denne samme teknikken, gitt de andre prosessen variablene styre frekvensen av gelation, frekvensen av electrospinning og frekvensen av fordampning under electrospinning forblir uendret.
Interessant, avhengig av hvilken metode som brukes til å forberede de forskjellige morphologies, har betydelige forskjeller blitt observert i dårligere tider hydrogels forberedt fra samme hydrogel forløpere. For eksempel redusere POEGMA nanofibrous hydrogels tregere enn bulk POEGMA hydrogels med samme sammensetning til tross for sin betydelig høyere areal og dermed tilgang til vann til hydrolyze hydrazone obligasjoner. Vi forholde seg disse forskjellene iboende kontrastene mellom beskrevet protokollene i geometri blande forløper polymerer, som kan føre til interne gel homogeneities og/eller morphologies som er vesentlig forskjellig og/eller i situ konsentrasjon av polymer forløpere på samme tidsskalaen som gelation, særlig relevant i electrospinning samtidige fordampning og crosslinking observert i denne prosessen. Mens dette kan noe komplisere valg av forløperen polymerer hvis en polymer er beregnet for bruk i hver protokoll, kan det også tilby en teknisk mulighet i å gjøre hydrogels med en kjemisk sammensetning, men svært forskjellige fysiske egenskaper.
Samlet av metodene beskrevet gir en strategi for fabrikasjon nedbrytbart (eller minst renally clearable) analoger av thermoresponsive polymerer på flere lengde skalaer (bulk, mikro og nano) og med flere typer interne strukturer (partikler eller fiber). Slike protokoller ta nøkkelen barrierer til vellykket oversettelsen av konvensjonelt forberedt syntetisk thermoresponsive materialer til feltet biomedisinsk: injectability og nedbrytbarhet. Vi fortsetter å utforske anvendelsen av slike materialer i både narkotika-leveranser og vev utvikling programmer alt fra det fysiske mål av kreft, transport av narkotika over blod – hjerne barrieren, terapeutiske levering av proteiner på baksiden av øyet, retningsbestemt vekst av vev, og thermoreversible vedheft og differensiering av celler, blant andre programmer.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
Finansiering fra naturvitenskap og Engineering Research Council for Canada (NSERC), NSERC Opprett-IDEM (integrert Design av ekstracellulære matriser) programmet, 20/20: NSERC ophthalmica biologisk materiale Research Network og Ontario departementet for forskning og Innovasjon tidlig forsker utmerkelser programmet er anerkjent.
Materials
| Chemicals | |||
| 2,2 – azobisisobutryic acid dimethyl ester | Wako Chemicals | 101138 | |
| Di(ethylene glycol) methyl ether methacrylate (M(EO)2MA) | Sigma Aldrich | 447927 | 188.2 g/mol, n=2 ethylene oxide repeat units |
| Oligo (ethylene glycol) methyl ether methacrylate (OEGMA475) | Sigma Aldrich | 447943 | 475 g/mol, n=8-9 ethylene oxide repeat units |
| Acrylic acid (AA), 99% | Sigma Aldrich | 147230 | |
| Thioglycolic acid (TGA), 98% | Sigma Aldrich | T3758 | |
| Dioxane, 99% | Caledon Labs | 360481 | |
| Nitrogen, UHP grade | Air Liquide | Alphagaz1 765A-44 | |
| Adipic acid dihydrazide (ADH), 98% | Alfa Aesar | A15119 | |
| N'-ethyl-N-(3- dimethylaminopropyl)-carbodiimide (EDC, x%) | Carbosynth | FD05800 | |
| Hydrochloric acid (HCl), 37% | Sigma Aldrich | 320331 | |
| Sodium hydroxide (NaOH), 97% | Sigma Aldrich | 221465 | |
| Aminoacetyl aldehyde dimethyl acetal, 99% | Sigma Aldrich | 121967 | |
| 4-Hydroxy-TEMPO, 97% | Sigma Aldrich | 176141 | |
| Methacryloyl chloride,97x% | Sigma Aldrich | 523216 | |
| Petroleum ether, 95% | Sigma Aldrich | 32047 | |
| Magnesium sulfate, 99.5% | Sigma Aldrich | M7506 | |
| tert-Butyl methyl ether, >99.0% | Sigma Aldrich | 443808 | |
| Phosphate buffered saline | BioShop | PBS405.1 | 1x, pH 7.3-7.5 |
| N-isopropylacrylamide, 99% | J&K Scientific | 258717 | Recrystallized from 60% hexanes/40% toluene |
| Ethanol, anhydrous | Commerical Alchols | P016EAAN | |
| Span 80 | Sigma Aldrich | S6760 | |
| Heavy paraffin oil | Caledon Labs | 1326197 | |
| Pentane, reagent grade | Caledon Labs | 1/10/7800 | |
| Poly (ethylene oxide) average Mv 600,000 | Sigma Aldrich | 182028 | |
| Supplies essential for synthesis and hydrogel fabrication | |||
| Rotary evaporator | Heidolph | G3 | |
| Dialysis tubing (3500 Da molecular weight cut-off) | Spectrum Labs | 28170-166 | Vol/length= 6.4mL/cm |
| Double barrel syringe | Medmix | L series | L series, 2.5 mL, 1:1 volume ratio |
| Static mixer | Medmix | L series | L series, 2.5 mL, 1:1 volume ratio, 1.5" length |
| Silicone rubber sheet, 1/16" thickness | McMaster-Carr | 9010K12, 30A Durometer (Super Soft) | |
| Syringe pump | KD Scientific | KDS Legato 200 | Infuse Only Dual Syringe Pump |
| High voltage power supply | Spellman | 230-20R | 0 to 20 kV |
| Microfluidic Chip Fabrication | |||
| Silicon wafer | University Wafer | 2080 | D = 76.2 mm; 380 µm thickness; P-doped; <100> orientation |
| SU-8 100 | MicroChem | Y131273 | |
| SU-8 Developer | MicroChem | Y020100 | |
| Custom 2.5" spincoater | Built in-house | N/A | |
| Mask Aligner | KARL SUSS | MJB3 UV400 (with a 276 W lamp) | |
| Masterflex L/S 13 Silicone Tubing | Cole Parmer | OF-96400-13 | Peroxide-cured |
| Dow Corning Sygard 184 Silicone Elastomer Base | Ellsworth Adhesives | 4019862 | |
| Dow Corning Sygard 184 Silicone Elastomer Curing Agent | Ellsworth Adhesives | 4019862 | |
| High Power Plasma Cleaner | Harrick | PDC-002-HP | |
| Characterization Instruments | |||
| Mach 1 micromechanical tester | Biomomentum | LB007-EN | |
| Cellstar tissue culture 12 well plate | Greiner Bio-one | 665 180 | |
| Cell culture insert for 12 well plate | Corning | 08-771-12 | 8 µm pore size |
| Optical microscope | Olympus BX51 optical microscope | BX51 | |
| Temperature-controlled microscope stage | Linkam Scientific | THMS600 | |
| Gel permeation chromatograph (GPC) | Waters | 590 HPLC Pump | Waters Styragel columns (HR2, HR3, HR4; 30 cm x 7.8 mm (ID); 5 mm particles), Waters 410 refractive index detector |
| Dynamic light scattering (DLS) | Brookhaven | 90Plus Particle Size Analyzer | |
| Transmission electron microscopy (TEM) | TEMSCAN | JEOL 1200EX | Accelerating voltage 100 kV |
| Scanning electron microscopy (SEM) | Tescan | Vega II LSU | Accelerating voltage 10 kV |
| Microsquisher | CellScale Biomaterials Testing | MS-50M-01 |
Riferimenti
- Heskins, M., Guillet, J. E. Solution Properties of Poly(N-isopropylacrylamide). J. Macromol. Sci. A. 2 (8), 1441-1455 (1968).
- Lutz, J. -. F., Akdemir, &. #. 2. 1. 4. ;., Hoth, A. Point by Point Comparison of Two Thermosensitive Polymers Exhibiting a Similar LCST: Is the Age of Poly(NIPAM) Over. J. Am. Chem. Soc. 128 (40), 13046-13047 (2006).
- Pelton, R. H., Chibante, P. Preparation of Aqueous Lattices with N-Isopropylacrylamide. Colloids Surf. 20 (3), 247-256 (1986).
- Palasis, M., Gehrke, S. H. Permeability of Responsive Poly(N-Isopropylacrylamide) Gel to Solutes. J. Controlled Release. 18 (1), 1-11 (1992).
- Kawaguchi, H., Fujimoto, K., Mizuhara, Y. Hydrogel Microspheres .3. Temperature-Dependent Adsorption of Proteins on Poly-N-Isopropylacrylamide Hydrogel Microspheres. Colloid Polym. Sci. 270 (1), 53-57 (1992).
- Okuyama, Y., Yoshida, R., Sakai, K., Okano, T., Sakurai, Y. Swelling Controlled Zero-Order and Sigmoidal Drug-Release from Thermoresponsive Poly(N-Isopropylacrylamide-Co-Butyl Methacrylate) Hydrogel. J. Biomater. Sci. Polym. Ed. 4 (5), 545-556 (1993).
- Snowden, M. J. The Use of Poly(N-Isopropylacrylamide) Lattices as Novel Release Systems. J. Chem. Soc. – Chem. Comm. (11), 803-804 (1992).
- Haraguchi, K., Takehisa, T., Ebato, M. Control of cell cultivation and cell sheet detachment on the surface of polymer/clay nanocomposite hydrogels. Biomacromolecules. 7 (11), 3267-3275 (2006).
- Lee, B., et al. Initiated chemical vapor deposition of thermoresponsive poly(N-vinylcaprolactam) thin films for cell sheet engineering. Acta Biomater. 9 (8), 7691-7698 (2013).
- Cole, M. A., Voelcker, N. H., Thissen, H., Griesser, H. J. Stimuli-responsive interfaces and systems for the control of protein-surface and cell-surface interactions. Biomaterials. 30 (9), 1827-1850 (2009).
- Feil, H., Bae, Y. H., Feijen, J., Kim, S. W. Molecular Separation by Thermosensitive Hydrogel Membranes. J. Membrane Sci. 64 (3), 283-294 (1991).
- Kim, J., Park, K. Smart hydrogels for bioseparation. Bioseparation. 7 (4-5), 177-184 (1998).
- Yamashita, K., Nishimura, T., Nango, M. Preparation of IPN-type stimuli responsive heavy-metal-ion adsorbent gel. Polym. Adv. Tech. 14 (3-5), 189-194 (2003).
- Ziolkowski, B., Czugala, M., Diamond, D. Integrating stimulus responsive materials and microfluidics: The key to next-generation chemical sensors. J. Intelligent Mater. Syst. Struct. 24 (18), 2221-2238 (2013).
- Zhang, Y., Kato, S., Anazawa, T. A flap-type hydrogel actuator with fast responses to temperature. Smart Mater. Struct. 16 (6), 2175-2182 (2007).
- Suzuki, D., Taniguchi, H., Yoshida, R. Autonomously Oscillating Viscosity in Microgel Dispersions. J. Am. Chem. Soc. 131 (34), 12058-12059 (2009).
- Schild, H. G. Poly(N-isopropylacrylamide): Experiment, Theory and Application. Prog. Polym. Sci. 17, 163-249 (1992).
- Oh, J. K., Min, K., Matyjaszewski, K. Preparation of poly (oligo (ethylene glycol) monomethyl ether methacrylate) by homogeneous aqueous AGET ATRP. Macromolecules. 39 (9), 3161-3167 (2006).
- Vihola, H., Laukkanen, A., Tenhu, H., Hirvonen, J. Drug Release Characteristics of Physically Cross-Linked Thermosensitive Poly(N-vinylcaprolactam) Hydrogel Particles. J. Pharm. Sci. 97 (11), 4783-4793 (2008).
- Zhang, L. F., Liang, Y., Meng, L. Z. Thermo-sensitive amphiphilic poly(N-vinylcaprolactam) copolymers: synthesis and solution properties. Polym. Adv. Tech. 21 (10), 720-725 (2010).
- Smeets, N. M. B., Bakaic, E., Patenaude, M., Hoare, T. Injectable and tunable poly(ethylene glycol) analogue hydrogels based on poly(oligoethylene glycol methacrylate). Chem. Comm. 50 (25), 3306-3309 (2014).
- Lutz, J. -. F. Polymerization of oligo (ethylene glycol)(meth) acrylates: toward new generations of smart biocompatible materials. J. Polym. Sci. A. 46 (11), 3459-3470 (2008).
- Lutz, J. -. F., Hoth, A. Preparation of Ideal PEG Analogues with a Tunable Thermosensitivity by Controlled Radical Copolymerization of 2-(2-Methoxyethoxy)ethyl Methacrylate and Oligo(ethylene glycol) Methacrylate. Macromolecules. 39 (2), 893-896 (2006).
- Patenaude, M., Campbell, S., Kinio, D., Hoare, T. Tuning Gelation Time and Morphology of Injectable Hydrogels Using Ketone-Hydrazide Cross-Linking. Biomacromolecules. 15 (3), 781-790 (2014).
- Patenaude, M., Hoare, T. Injectable, Degradable Thermoresponsive Poly(N-isopropylacrylamide) Hydrogels. ACS Macro Lett. 1 (3), 409-413 (2012).
- Patenaude, M., Hoare, T. Injectable, Mixed Natural-Synthetic Polymer Hydrogels with Modular Properties. Biomacromolecules. 13 (2), 369-378 (2012).
- Smeets, N. M. B., Bakaic, E., Patenaude, M., Hoare, T. Injectable poly(oligoethylene glycol methacrylate)-based hydrogels with tunable phase transition behaviours: Physicochemical and biological responses. Acta Biomater. 10 (10), 4143-4155 (2014).
- Sivakumaran, D., Mueller, E., Hoare, T. Temperature-Induced Assembly of Monodisperse, Covalently Cross-Linked, and Degradable Poly(N-isopropylacrylamide) Microgels Based on Oligomeric Precursors. Langmuir. 31, 5767-5778 (2015).
- Bakaic, E., Smeets, N. M. B., Dorrington, H., Hoare, T. “Off-the-shelf” thermoresponsive hydrogel design: tuning hydrogel properties by mixing precursor polymers with different lower-critical solution temperatures. RSC Adv. 5 (42), 33364-33376 (2015).
- Bulpitt, P., Aeschlimann, D. New strategy for chemical modification of hyaluronic acid: Preparation of functionalized derivatives and their use in the formation of novel biocompatible hydrogels. J. Biomed. Mater. Res. 47 (2), 152-169 (1999).
- Kesselman, L. R. B., Shinwary, S., Selvaganapathy, P. R., Hoare, T. Synthesis of Monodisperse, Covalently Cross-Linked, Degradable “Smart” Microgels Using Microfluidics. Small. 8 (7), 1092-1098 (2012).
- Sivakumaran, D., Mueller, E., Hoare, T. Microfluidic production of degradable thermoresponsive microgels based on poly(N-isopropylacrylamide). Soft Matter. , (2016).
- Xu, F., Sheardown, H., Hoare, T. Reactive Electrospinning of Degradable Poly(oligoethylene glycol methacrylate)-Based Nanofibrous Hydrogel Networks. Chem. Comm. 52 (7), 1451-1454 (2016).
- Troll, K., et al. The collapse transition of poly(styrene-b-(N-isopropyl acrylamide)) diblock copolymers in aqueous solution and in thin films. Colloid Polym. Sci. 286 (8), 1079-1092 (2008).
- Patenaude, M., Campbell, S., Kinio, D., Hoare, T. Tuning Gelation Time and Morphology of Injectable Hydrogels Using Ketone-Hydrazide Cross-Linking. Biomacromolecules. 15 (3), 781-790 (2014).
- Kelly, T. A., Felder, M. S., Ollar, R. A. Inducing Apoptosis in a Mammalian Cell by Contacting with Paraffin or Agar. US Patent. , (2001).
