


ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
クラスター化された定期的に空間短い回文の繰り返し/CRISPR 関連付けられている蛋白質 9 (CRISPR/Cas9) システムは遺伝子工学の有望なツールを提供し、transgenes の対象となる統合の可能性を開きます。(HMEJ) に参加する相同性の終わりについて述べる-ベースの戦略を効率的な DNA が生体内で統合対象し、標的遺伝子療法 CRISPR/Cas9 を使用しています。
Abstract
有望なゲノム編集プラットフォームとして CRISPR/Cas9 システム特に遺伝子の目標とされた統合のための効率的な遺伝的操作のための大きな可能性があります。しかしながら、相同組換え (HR) の効率が低い、非相同末端結合 (NHEJ) 様々 な塩基変異の非分裂細胞、生体内でゲノム編集のままで戦略に大きな課題をに基づいて。(HMEJ) に参加する相同性の終わりを述べる-ベースの正確なターゲットを絞った統合効率的な体内の CRISPR/Cas9 システム。本システムで対象となるゲノムやドナー ベクトル含むホモロジー腕 (~ 800 bp) シーケンスは CRISPR/Cas9 によって裂かれる単一ガイド RNA (sgRNA) ターゲットが並ぶ。この HMEJ ベース作戦マウス受精卵だけでなく、生体内の肝細胞の効率的な遺伝子の統合を実現します。また、HMEJ ベースの戦略の肝細胞における 4-フマリルアセト酢酸加水分解酵素 (ファー) 突然変異の補正のための効率的なアプローチを提供しています、 Fahを救う-欠乏誘発肝障害マウス。一緒に取られて、統合をターゲットに焦点を当てて、この HMEJ ベースの戦略は、有望な世代の遺伝子組み換え動物モデルおよび目標とされた遺伝子治療を含む、アプリケーションのさまざまなツールを提供します。
Introduction
正確なターゲットを絞ったゲノム編集がしばしば遺伝子組み換え動物モデルや臨床治療必要です。効率的な標的ゲノム ジンクフィンガーヌクレアーゼ (ZFN) など、転写活性化因子のようなエフェクター核酸分解酵素 (TALENs) を編集のため様々 な戦略を開発する多くの努力をなされている、CRISPR/Cas9 システム。これらの戦略はゲノムのターゲット DNA 二重鎖切断 (DSB) を作成し、相同組換え (HR)1,2, microhomology を介した終了 (MMEJ)3に参加するなど、本質的な DNA 修復システムを活用,4,5、および非相同末端結合 (NHEJ)6,7,8 transgenes1,9の対象となる統合を誘発します。人事戦略は現在最も一般的に使用されるゲノム編集アプローチは、細胞株で非常に効率的なが、後半の S/G2 段階でその制限の発生による非分裂細胞に容易にアクセスできません。したがって、人事戦略は生体内でゲノム編集のため適用されません。最近、NHEJ ベースの戦略は、マウス組織8での効率的な遺伝子ノックアウトのために開発されました。それにもかかわらず、NHEJ 方式は通常オクターリピート困難に正確なゲノムの編集、特にフレームの融合遺伝子8を構築しようとしたときに生成する交差点を紹介します。MMEJ ベースの対象となる統合は正確な編集が可能です。ただし、のみ控えめ前レポート5の対象となる統合効率が高まります。したがって、生体内で正確なターゲットを絞った統合の効率の向上が急務幅広い治療への応用3。
最近出版された仕事で行った (HMEJ) に参加する相同性の終わり-ベースの戦略は、報告されたすべての戦略を両方生体外でそして生体内の10の最高のターゲットを絞った統合効率を示した。ここでは、HMEJ システムの確立のためのプロトコルについて述べるし、関心とドナーの遺伝子をターゲット シングル ガイド RNA (sgRNA) ベクトルの建設ベクトル遺伝子 sgRNA のターゲット ・ サイトと 〜 800 も相同性腕 (図 1) の bp.このプロトコルでも世代 DNA ノックイン マウスと簡単な手順の生体内の組織の目標の統合のための詳細な手順を説明します。さらに、HMEJ ベースの戦略の概念の実証研究は、 Fah変異を修正し、救助のFah-/-肝障害マウス、さらにその治療の可能性を明らかにする能力を示した。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
動物科目を含むすべてのプロシージャは、生物科学 (CAS) の上海研究所の生物医学研究倫理委員会で承認されています。
1. ドナー プラスミドのデザイン
-
SgRNA の選択
- オンライン CRISPR デザイン ツールを使用して、ターゲット地域11,12,13,14,15sgRNAs を予測します。Cdx2遺伝子座で、六つの異なる sgRNAs の設計 (Cdx2-sgRNA1 ~Cdx2-sgRNA6) 停留所周辺より高いランクと下限のポテンシャルを持つコドンをオフ-ターゲット (図 1A)16。
- BbsI 消化 (1 U/μ L で 20 μ L のボリュームの最終濃度 37 ° C で 2 時間に BbsI の 1 μ L) Cas9 CMV EGFP 発現ベクターと sgRNA の 2 μ g をリニア化します。1 × TAE バッファーに 1% の agarose のゲルのゲル精製キットによって浄化製品。
- ミックスを 50 μ M の最終濃度 1 × T4 DNA リガーゼ バッファーの 10 μ L の sgRNA オリゴヌクレオチドのペア孵化 95 ° C から 25 ° C への温度勾配を用いた温度変化率 5 ° C/5 分 (5 分の 95 ° C オリゴ ソリューション、5 分、 5 分などの 85 ° C、90 ° C.)、oligos をアニールするでしょう。
- ミックス 4 μ L の焼鈍製品、1 μ L 1 × T4 DNA リガーゼ バッファーの 10 μ L の T4 DNA リガーゼの一直線に並べられたベクトルの 2 μ L と、1-2 h (図 1B) 22 ° C で縛る。
-
SgRNA の測量ヌクレアーゼ アッセイ
注: 測量ヌクレアーゼ アッセイによってノックアウトの実験に使われて sgRNA のターゲット効率を評価 (として知られている T7 エンドヌクレアーゼ I (T7EI) アッセイ)17。高い DNA の開裂の効率と低距離 sgRNA 切断サイト停止コドンと、sgRNA を選択します。- N2a セルラインに Cas9 sgRNA EGFP 発現ベクターを transfect 10% 牛胎児血清を DMEM 培養、1 %psg、および 1% 非必須アミノ酸で、トランスフェクション キット (材料の表を参照してください)。5% CO2の 37 ° C で transfected セルを孵化させなさい。
- 培養 48 時間後収集蛍光活性化セル (FACS) を並べ替えによって 5,000 transfected セル (GFP+) コントロールとして非 transfected セルを使用します。
- 2-5 56 ° C、30 分で oflysis バッファーを μ L (0.1% トリトン X-100, 0.1% Tween 20、および 100 μ g/mL プロティナーゼ K) で収集された細胞を消化し、熱が 95 ° c 10 分プロテイナーゼ K を不活性化します。
- Nested pcr 法 (表 1) の製造元のプロトコルを使用して、サンプルを増幅します。PCR の製品のサイズは、300-500 bp に設定されます。
- ミックス 1 μ L の DNA ポリメラーゼと sgRNA ターゲット サイト (0.1 μ M、最終濃度) をシーケンスを認識し外側のプライマーのペアを持つ溶解製品 (表 1)、20 μ L のボリュームのプライマリの PCR を行ったりします。
- 95 ° C、5 分で DNA ポリメラーゼをアクティブ化して 30 95 ° C で 30 サイクルの主要な PCR を実行 s、60 ° C、30 s、および 24 のための 72 ° C s (1 分/1 kb)、5 分のための 72 ° C で最終的な拡張子を持つ。
- 主要な PCR の製品の 1 μ L と入れ子になった内側のプライマーのペアを使用してセカンダリ PCR を実行します。
- 変性し、再アニール 1 × T7EI 反応バッファー (50 mM NaCl、10 mM 10 mM MgCl2、トリス塩酸 1 mM DTT pH 7.9) の 20 μ L の精製 PCR の製品の 300-600 ng 率が 5 ° C/5 分の 95 ° C から 25 ° C への温度勾配を使用して。
- 焼なましの PCR の製品と 2 h の 37 ° C でダイジェストに T7EI 酵素の 1 μ L を追加します。(材料の表を参照してください) フラグメントを分離するまでは、40 分の 120 V で 1 × TAE バッファーに 2% の agarose のゲルで消化製品を実行します。
- ImageJ を使用して、カットとカットされていない DNA のバンドの強度を決定します。前述の方法を使用して塩基頻度報告9 (図 1C) を計算します。
-
ドナー ベクターの構築
注: Cdx2遺伝子の HMEJ ドナー ベクトルを生成する構築ドナー DNA (800 bp HAL p2A-mCherry 800 bp HAR) の両脇に 23 nt Cdx2 sgRNAs ターゲット シーケンス (図 1Dと図 1E) 両端。ターゲット シーケンスの PAM は相同のアームの先端に隣接してだった。HMEJ ドナーのクローン作成にはギブソン アセンブリをお勧めします。- 前方のプライマーを 1 階で 800 bp 左側相同アーム (HAL) の増幅 (ベクトル、23 から 15-20 nt 重複シーケンスを含む nt Cdx2 sgRNA ターゲット シーケンス、および HAL から約 20 nt シーケンス) と逆プライマー-1R (から 15-20 bp 重複シーケンスを含むp2A mCherry と HAL から約 20 nt シーケンス) 200 ng/μ L (表 1図 1D) でマウス genomic DNA を用いた 0.1 μ M 最終濃度で。
- 前方プライマー 2 階 (HAL から 15-20 nt 重複シーケンスおよび挿入フラグメントから約 20 nt シーケンスを含んでいる) で p2A mCherry 挿入断片の増幅、逆プライマー-2R (HAR から 15-20 nt 重複シーケンスおよびからの約 20 の nt シーケンスを含んでいます。挿入断片) 0.1 μ M 最終濃度 100 ng/μ L または 30 ng/μ L (表 1図 1D) で記者シーケンスでゲノム DNA やプラスミドを使用します。
- 前方プライマー 3F で 800 bp 右側相同アーム (HAR) の増幅 (ベクトル、23 から 15-20 nt 重複シーケンスを含む nt Cdx2 sgRNA ターゲット シーケンスとハーから約 20 nt シーケンス) と逆プライマー-3R (から 15-20 nt 重複シーケンスを含むp2A mCherry ・ ハーから約 20 nt シーケンス) 200 ng/μ L (表 1図 1D) でマウス genomic DNA を用いた 0.1 μ M 最終濃度で。
- 1 × TAE バッファーに 1% の agarose のゲルの PCR の製品をすべてを実行し、製造元の指示 (表 1) によるとゲル抽出用のキットによって予想されるサイズの PCR の製品を浄化します。
- KpnI と XbaI 構造ベクトルの 50-100 ng を消化します。30 40 ng/μ L と 3 つの PCR で一直線に並べられたベクトルのミックス 2 μ L は、ギブソン ミックス × 2 で (各、100-200 ng/μ L の 1 μ L) の断片を増幅しました。H2O 10 µL.Incubate 50 ° C, 60 分でミックスする最終的な音量を調整するを追加します。
- 有能なエシェリヒア属大腸菌を変形すべての組み立て製品とエキスの製造元の指示に従って DNA 抽出キットによってプラスミッドを構築します。HMEJ ドナー DNA 塩基配列を確認します。
2. ゲノム HMEJ 法を用いたマウス胚での編集
-
Cas9 mRNA の生産
- Cas9 mRNA の準備、表 1に記載されている適切なプライマー対を用いた PCR 法で地域のコーディング、Cas9 に T7 プロモーターのシーケンスを追加します。プライマー Cas9 F/R を追加最終濃度 0.1 μ M と 20 で 1 × 高忠実度 DNA ポリメラーゼ ミックスへのベクトルを表現する Cas9 の ng。H2O を 50 μ L に最終的な音量を調整します。
- 95 ° C、5 分で DNA ポリメラーゼをアクティブ化して 30 95 ° c 36 サイクルの PCR を実行 s、60 ° C、30 秒と 4 分 (1 分/1 kb)、68 ° C 10 分で最後の拡張のための 68 の ° C。
- In vitro転写 (IVT) T7 Cas9 PCR の製品の浄化し、0.5-1 を書き写す μ g DNA の製造元の指示に従って、20 μ L の容量で 8 h の 37 ° C で mRNA 転写キット (材料の表を参照してください)。
- 15 分追加 37 ° C の 45 分のポリ A テールの 37 ° C で DNA テンプレートを削除し、製造元の指示に従って RNA 精製キットで Cas9 mRNA を回復する混合物に DNase の 1 μ L を追加 (材料の表を参照してください)。
-
SgRNA の生産
- 上記として忠実性の高い DNA ポリメラーゼと T7 プロモーターによって駆動 sgRNA テンプレートを生成します。テンプレートとしてベクターを含む sgRNA 足場を選択します。使用されるプライマーは、表 1に示します。
- T7 sgRNA PCR の製品の浄化し、0.5-1 を使って μ gの in vitro転写 sgRNA 37 ° C で製造元の指示に従って、20 μ L の総ボリュームで 6 時間の短い RNA 転写キットを使用してのためのテンプレートとしての DNA (材料の表を参照してください <。/c11 >)。
- 混合物に DNase の 1 μ L を追加し、DNA テンプレートを削除する 15 分の 37 ° C で培養を続けます。RNA 精製キット、浄化、sgRNAs (材料の表を参照してください) 上記のように。
- RNase フリー水 500 ng/μ L に sgRNA を希釈し、3 ヵ月の −80 ° C で試料を保存します。
注: CRISPR ribonucleoproteins (結合) より良い切削効率18,19,20を持つ代替置換です。
-
胚のコレクション、マイクロインジェクション、体外培養
- Superovulate 女性 B6D2F1 (C57BL/6 × DBA2J) (7-8 週古い) によるマウス妊馬血清性腺刺激ホルモン (PMSG)、48 時間後でひと絨毛性ゴナドトロピン (hCG) が続きます。HCG 注射後の家 B6D2F1 の男性と女性は一晩します。
- CO2麻酔、hCG 注射後 24 h によって女性を犠牲に。M2 中 (各女性のための 30-50 胚) とその卵管から受精卵を収集します。
- 場所 KSOM 中に受精卵 (1 日注射約 300 卵) (5.55 g/L の NaCl、KCl、0.05 g/L KH2PO4、0.05 g/L MgSO4•7H2O、0.04 g/L, グルコース 1.12 g/L ナトリウム 0.19 g/L 乳酸、2.1 g/L NaHCO3、ピルビン酸ナトリウム 0.02 g/L、0.25 g/L CaCl2•2H2O、0.004 g/L EDTA、0.146 g/L L-グルタミン 1 G/L のウシ血清アルブミン) で 5% CO2インキュベーター 37 ° C。
- ミックスの Cas9 mRNA (100 ng/μ L)、sgRNA (50 ng/μ L)、および HMEJ ドナー (100 ng/μ L) をベクトル H2O 10 μ L に最終的な音量を調整する氷の混合物を入れを追加。
- キャピラリー針 (外径 1.0 mm、内径フィラメント 0.78 mm) を引くマイクロ ピペットの引き手を使用して (パラメーター: 熱、74; プル、60; 速度 80; 200 時間/圧力、300。材料の表を参照)。商業の針をマイクロインジェクションの代替置換となります。
- 5 μ g/mL サイトカラシン B 定数とマイクロを使用してを含む HEPES CZB 中の液滴の明確に定義された前核と受精卵の細胞質に混合物のありそうなボリュームを挿入フロー設定 (図 2A) (を参照してください。材料のテーブル)21。
注: 受精卵の各グループを注入する必要があります投与後 20-30 分以内サイトカラシン B がマウス受精卵の生存率を高めることができます。また、マイクロインジェクションは22を前述のように圧電システムで操作できます。 - KSOM 培地で 37 ° C 未満 5%、胚盤胞まで CO2ステージ蛍光観察 (図 2 bおよび 2C) の 3.5 日後に挿入された受精卵を培養します。
-
胚移植とマウスの生成
- 注射と同じ日に発情 icr 系雄マウスの精管の icr 系雌マウスを仲間します。
- 5% の CO2と 0.5 日のポストの coitum (dpc) で偽の icr 系雌の卵管に移植 25 30 2 細胞胚下 37 ° C で 2 細胞期に注入された受精卵を培養します。受信者の母親は、19.5 dpc で子犬を提供します。
-
マウスのジェノタイピング
- 製造元の指示に従って DNA 抽出キットを使用してつま先または尾のサンプルからマウスのゲノム DNA を抽出 (材料の表を参照してください)。
- ゲノム DNA をテンプレートとして紫外/可視分光分析法による測定の 200-400 ng を使用して PCR させずに増幅を実行するノックアウトのイベントの 5' と 3' のジャンクションを識別します。
- 95 ° C、5 分で DNA ポリメラーゼをアクティブ化して 30 95 ° c 38 サイクルの PCR を実行 s、60 ° C、30 秒、1 分 (1 分/1 kb)、72 ° C 10 分で最後の拡張のための 72 ° C。5' ジャンクション、逆ノックイン フラグメント (p2A-mCherry) で、HAL の上流で前方のプライマーを使用します。3' ジャンクション、に関して HAR (表 1) の下流に、逆に 1 でノックのフラグメント (p2A-mCherry) の前方のプライマーを使用します。
- 1 × TAE バッファー、および予想されるフラグメントのサイズのチェックに 1% の agarose のゲルの PCR の製品の 6 μ L を実行します。(図 2D) を配列する DNA によってそれらを確認します。
3. HMEJ ベースはIn Vivoゲノム肝細胞での編集
- 拘束デバイスで受信者の c57bl/6 j マウス (8 週) を置き、尾をスリットに通します。
- HMEJ ドナー ベクトル (30 μ g) と spCas9 発現ベクター (30 μ g) 食塩液 2 ml を混ぜます。コントロール実験のため食塩 (図 3A) の 2 mL で HMEJ ドナー ベクトル (30 μ g) は中止します。
- 70% エタノールでねずみのしっぽをクリーンアップします。尾に針が静脈内 5-7 s. プラスミド DNA 溶液を注入して挿入針を除去し、抑制デバイスからマウス ボタンを離します。
- CO2麻酔によって注入後 5-9 日後マウスを犠牲に。マウス transcardially 0.9% 生理食塩水を灌流、ペリスタ ポンプ 4% パラホルムアルデヒドに続いて、4 ° C で一晩肝臓を修正
- チューブの底に沈むまで一晩、30% ショ糖を使用して組織を脱水します。
- 肝臓サンプルの 10 μ m の厚さで、凍結するティッシュをセクションします。
- すすぎ 3 回で 0.1 M のリン酸バッファー (PB) セクションとそれらを一次抗体とインキュベート: ウサギの抗 mCherry (5% で希釈した NGS) 4 ° C でオーバー ナイト
- PB の 3 回のセクションを洗浄し、二次抗体とのそれらを孵化させなさい: Cy3 AffiniPure ヤギ抗うさぎ IgG 軌道シェーカーで室温で 2 時間。
- 対比染色 20 分 DAPI セクションとさらに蛍光観察 (図 3B) のスライド ガラスのグリセリンでマウント。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
マウス胚における HMEJ を用いたゲノム編集:マウス受精卵の HMEJ 方式のノックの効率を定義するには、納品 Cas9 mRNA、 Cdx2 の最後のコドンに p2A mCherry レポーターの遺伝子を融合するように設計されたマウスの受精卵へCdx2遺伝子や HMEJ ドナーを対象とした sgRNA遺伝子 (図 2A)。文化で胚盤胞に注入された受精卵を開発しました。蛍光顕微鏡を用いた mCherry 蛍光ノックの効率を評価する分析したし、栄養外胚葉で厳密に表現された mCherry の受信 HMEJ ドナー胚盤胞の 12.9% が陽性がわかった (図 2B、2 C)。PCR の積極的なマウスを配列することによっては、5' と 3' の両方の接合 (図 2D) 正確なフレームの統合は、統合イベントを調べてすべてことを発見しているも。
HMEJ ベース ゲノム編集成体組織と HMEJ 媒介性遺伝子治療:成体組織における HMEJ を用いたゲノム編集を適用できるかどうか調べるために、尾静脈で C57/B6J マウス肝臓Actb- HMEJ 構造を伝達によってActb遺伝子の終止コドン直前 mCherry カセットを挿入流体注入 (図 3A)。注射 7 日後肝切片 (図 3B) 上のステンド グラスとしては、mCherry を表現 transfected 肝細胞のほぼ半分で、ことがわかった。
4-フマリルアセト酢酸加水分解酵素 (ファー) を用いて、遺伝子治療用 HMEJ ベースの戦略を使用しての可能性を探検する-欠損マウス。Fahの-/-マウスは、確立された遺伝性 tyrosinemia 型 (HTI) マウス モデルは、次のシーケンスの23フレーム シフト突然変異を引き起こして、 Fah遺伝子の exon 5 挿入断片の港湾です。Fahの-/-マウスを維持するために 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC)24チロシン異化経路の上流の阻害剤とFahの-/-マウスを扱われます。ここで MMEJ と HMEJ を介した遺伝子補正がFahの-/-マウスのFahの突然変異を救うことができるかどうかを参照してくださいに着手しました。我々 は力学 Cas9 構造を注射したFahMMEJ またはエクソン イントロンFah遺伝子の 4 に 5 を 14 のFahの cDNA を挿入するように設計、 Fah- HMEJ 構造と共にFah -/-マウスの肝臓 (図 3C). 投与後 1 週間 NTBC 肝臓の損傷 (図 3C) を誘導するために撤回されました。NTBC、 Fahの撤退後- FahHMEJ と Cas9 構造体を受け取るFah-/-マウスの修正された肝細胞は MMEJ 方式 (図 3Dよりもより効果的な増殖を示した).

図 1: HMEJ を介した体外統合対象と。
(A) sgRNAs の選択のための実験手法: 六つの異なる sgRNAs (Cdx2-sgRNA1 ~Cdx2-sgRNA6) より高いランクとターゲットを可能性のあるCdx2遺伝子座の終止コドンの周りされた選ばれたオンライン CRISPR デザインに基づいてツールです。Protospacer 隣接するモチーフ (PAM) シーケンスは赤です。(B) 実験設計: sgRNA、Cas9、および GFP を表現する、Cas9 の CMV-GFP 発現プラスミド N2a 細胞に導入されました。GFP+細胞は、測量士試験の 3 日目でソートされます。(C) 測量アッセイCdx2の対象: 6 の異なる sgRNAs 測量士試験用に設計されました。通常 N2a 細胞のゲノム DNA は、コントロールとして機能します。*、 Cdx2用 sgRNA-2 a-mCherry ノックアウトの実験。(D) HMEJ ドナー ギブソン アセンブリを使用しての構築の図式的な概観。(E) HMEJ を介した遺伝子Cdx2遺伝子座で戦略の図式的な概観。HAL/HAR、左/右の相同の腕;三角形、sgRNA ターゲット ・ サイト。/または、外側のフォワード/リバースのプライマー。場合/IR、内側正転/逆転のプライマー。図以前のレポート10から変更します。この図の拡大版を表示するのにはここをクリックしてください。

図 2: 統合を対象としたゲノム編集を介して HMEJ を介したマウス胚における
(A) 実験マイクロインジェクション法: Cas9 mRNA の混合物 (100 ng/μ L)、sgRNA (50 ng/μ L) とドナーのプラスミド (100 ng/μ L) は、マウスの受精卵に注入しました。(B) 代表的な蛍光画像編集 HMEJ 戦略によってマウス胚の。バー、mCherry+の割合で胚盤胞に示されている 20 μ m (C) ノックイン効率。上記の各バーは、カウント合計胚盤胞数です。(D) Cdx2遺伝子座遺伝子編集マウスの解析。PCR の製品、5' と 3' のジャンクション サイトから増幅されました。アッパー、ホモロジー アーム;紫、p2A;赤、mCherry;ハーまたは HAL、右側または左側相同腕。破線は、わかりやすくするため省略すると領域をマークします。図以前のレポート10から変更します。この図の拡大版を表示するのにはここをクリックしてください。

図 3: HMEJ を介した体内の統合対象と。
(A) 流体尾静脈注射の図式的な概観。ドナー シーケンスと sgRNA を表現するプラスミッドと spCas9 を表現するプラスミドの混合物は、流体の尾静脈注射を介して肝臓に届けられました。(B) 肝細胞の代表的な蛍光画像。肝のセクションでは、収集した 7 日のポスト噴射をだった。スケール バー、50 μ m. GFP は、細胞を導入しました。(C) プラスミドのいずれか MMEJ または HMEJ を介した遺伝子交換戦略 4 Fah遺伝子のイントロン ・ エクソン 5 に 14 のFah cDNA に挿入するように設計は、流体注入によるFah-/-マウスの肝臓に渡されました。上 NTBC: Fah-/-マウスは NTBC 水で維持されていたオフ NTBC: NTBC 水の回収 (NTBC 撤退の最初の日は注射後 7 日ある日 0 として定義されます)。(D) Fah MMEJ または HMEJ プラスミドを注入したFahの-/-マウスの肝切片の免疫組織化学染色します。スケール バー、100 μ m。 図は、以前のレポート5,10から変更。この図の拡大版を表示するのにはここをクリックしてください。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
HMEJ ドナー プラスミドの構造の最も重要な手順です: (1) 高 DNA の開裂の効率と低 sgRNA 切断サイト停止コドンと (2) 適切な建設 HMEJ ドナーの距離 sgRNA の選択。(SgRNA のターゲット ・ サイトと 〜 800 bp ホモロジー腕含む) 両方の transgene ドナー ベクトル CRISPR/Cas9 を介した胸の谷間、標的ゲノムは効率的かつ正確なターゲットを絞った統合に必要な体内。HMEJ 法を用いたノックアウトのマウスの世代の最も重要な手順は、: (1) 高品質の Cas9 mRNA と sgRNA (Cas9 mRNA および sgRNA の変性が存在しない) の準備をし、(2) 高品質 HMEJ ドナー プラスミッドの準備。プラスミッドは胚に有毒な効果を示しています。
最近、NHEJ 方式は効率的な体内ゲノム編集8のも報告されていた。それにもかかわらず、前レポート8正確な統合を達成するために難しく説明するよう各種塩基変異が通常接点で誘導された.ここでは、上に説明した HMEJ ベースの戦略は、ほとんど塩基変異の正確なターゲットを絞った統合を示した。したがって、HMEJ ベースの戦略は、NHEJ ベース方法は正しいものと (点突然変異) などの変異シーケンスを置換するための理想的なプラットフォームかもしれない。
モザイクは胚における遺伝子編集にとって大きな問題です。以前の萌芽期の段階で Cas9 mRNA ではなくタンパク質の注入は、モザイクなしの 1 つのセル段階での遺伝子ノックアウトの達成するかもしれない。臨床応用、成体に CRISPR/Cas9 システムの配信はまだ挑戦しています。
HMEJ に基づくゲノム編集の多くの将来の潜在的な用途があります。遺伝子組み換え動物モデルを生成する使用できます。そのノックの高性能胚を考慮したこのメソッドは遺伝子組み換え動物モデルを生成するために必要な動物の数を大幅に減らせるだろう、特に非ひと霊長類の遺伝学的モデルを生成する可能性を開きます。HMEJ に基づくゲノム編集できますリネージュ トレース個々 のセル型成体、ヒト以外の霊長類などの利用可能な動物モデルの欠乏があるので動物モデルに特に便利です。標的遺伝子療法のため使用することができます: HMEJ ベースの戦略の最も魅力的なアプリケーションは、クリニックでの遺伝子治療。本研究で我々 は遺伝性 tyrosinemia 型のFahの突然変異を修正私示されたベクトルの流体注入によるマウス。しかし、成体に CRISPR/Cas9 システムの配信は臨床使用の主要な技術的な挑戦でまだ流体注入は患者で実行される可能性があります。現在、さらに配信戦略の改善が急務クリニックにこの HMEJ 法を翻訳する前に。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者が明らかに何もありません。
Acknowledgments
この作品は CA 戦略的な重点研究プログラム (XDB02050007、XDA01010409)、国民のハイテク R & D プログラム (863 プログラム; 2015AA020307)、中国の国家自然科学基金によって支えられた (NSFC 助成金 31522037、31500825、31571509、31522038)、中国青少年千の才能プログラム (HY) に、ブレーク中国科学アカデミーのプロジェクトは、上海市科学技術委員会プロジェクト (ハイ、16JC1420202) は、科学省と中国の技術 (ほとんど; 2016YFA0100500)。
Materials
| Name | Company | Catalog Number | Comments |
| pX330 | Addgene | 42230 | |
| pAAV vector | Addgene | 37083 | |
| pX260 | Addgene | 42229 | |
| AAV_Efs_hSpCas9_NLS_FLAG-SV40 | Addgene | 97307 | AAV vector for encoding a human codon-optimized SpCas9 driven by EFs promoter |
| AAV_Actb HMEJ donor_U6_sgRNA_EF1a_GFP_polyA | Addgene | 97308 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Actb. EGFP driven by EF1a promoter and U6-driven sgRNAs targeting Actb. AAV backbone. |
| AAV_Cdx2 HMEJ donor | Addgene | 97319 | HMEJ donor for fusing a p2A-mCherry reporter to mouse Cdx2. |
| Lipofectamine 3000 Transfection Reagent | Life Technology | L3000015 | |
| Nuclease-Free Water | Life Technologies | AM9930 | |
| Bbs I | New England Biolabs | R0539S | |
| NEB Buffer 2 | New England Biolabs | B7002S | |
| T7 endonuclease I | New England Biolabs | M0302L | |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | E2621L | |
| Plasmid EndoFree-Midi Kit | Qiagen | 12143 | |
| MMESSAGE MMACHINE T7 ULTRA | Life Technologies | AM1345 | |
| MEGACLEAR KIT 20 RXNS | Life Technologies | AM1908 | |
| MEGASHORTSCRIPT T7 KIT 25 RXNS | Life Technologies | AM1354 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument | P-97 | Micropipette Puller (parameters: heat, 74; pull, 60; velocity, 80; time/delay, 200; pressure, 300) |
| Borosilicate glass | Sutter Instrument | B100-78-10 | type of capillaries (outer diameter 1.0 mm, inner diameter 0.78 mm with filament) |
| FemtoJet microinjector | Eppendorf | ||
| Freezing microtome | Leica | CM1950-Cryostat | thickness of 40 μm for brain, 10 μm for liver |
| Rabbit anti-mCherry | GeneTex | ||
| Cy3-AffiniPure Goat Anti-Rabbit IgG | Jackson Immunoresearch | ||
| DMEM | Gibco | 11965092 | |
| FBS | Gibco | 10099141 | |
| NEAA | Gibco | 11140050 | |
| Pen,Strep,Glutamine | Gibco | 10378016 | |
| Gel Extraction Kit | Omega | D2500-02 | |
| FACS | BD AriaII | ||
| PMSG | Ningbo Sansheng Medicine | S141004 | |
| HCG | Ningbo Sansheng Medicine | B141002 | |
| Cytochalasin B | Sigma | CAT#C6762 | |
| KSOM+AA with D-Glucose and Phenol Red | Millipore | CAT#MR-106-D | |
| M2 Medium with Phenol Red | Millipore | CAT#MR-015-D | |
| Mineral oil | Sigma |
References
- Yang, H., et al. Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154 (6), 1370-1379 (2013).
- Hockemeyer, D., et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 29 (8), 731-734 (2011).
- Nakade, S., et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nature Communications. 5, 5560 (2014).
- Hisano, Y., et al. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Scientific reports. 5, 8841 (2015).
- Yao, X., et al. Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine. , (2017).
- Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P., Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome research. 24 (1), 142-153 (2014).
- Maresca, M., Lin, V. G., Guo, N., Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Research. 23 (3), 539-546 (2013).
- Suzuki, K., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 540 (7631), 144-149 (2016).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Yao, X., et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Research. 27 (6), 801-814 (2017).
- Han, D. W., et al. Direct reprogramming of fibroblasts into epiblast stem cells. Nature Cell Biology. 13 (1), 66-71 (2011).
- Han, D. W., et al. Direct Reprogramming of Fibroblasts into Neural Stem Cells by Defined Factors. Cell Stem Cell. , (2012).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Sparman, M., et al. Epigenetic reprogramming by somatic cell nuclear transfer in primates. Stem Cells. 27 (6), 1255-1264 (2009).
- Schatten, G., Mitalipov, S. Developmental biology: Transgenic primate offspring. Nature. 459 (7246), 515-516 (2009).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Cong, L., et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339 (6121), 819-823 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18 (1), 92 (2017).
- Park, K. E., et al. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International journal of molecular sciences. 217 (6), (2016).
- Woo, J. W., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature biotechnology. 33 (11), 1162-1164 (2015).
- Harms, D. W., et al. Mouse Genome Editing Using the CRISPR/Cas System. Current protocols in human genetics. 83, 11-27 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature protocols. 9 (8), 1956-1968 (2014).
- Grompe, M., et al. Loss of Fumarylacetoacetate Hydrolase Is Responsible for the Neonatal Hepatic-Dysfunction Phenotype of Lethal Albino Mice. Genes & development. 7 (12), 2298-2307 (1993).
- Paulk, N. K., et al. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 51 (4), 1200-1208 (2010).
Tags
問題 133 CRISPR/Cas9、ターゲットを絞った統合、相同性を介した終わりに入社、生体内で胚の遺伝学遺伝子改変マウス、流体注入Erratum
Formal Correction: Erratum: CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy
Posted by JoVE Editors on 03/10/2021.
Citeable Link.
An erratum was issued for: Studying TGF-β Signaling and TGF-β-induced Epithelial-to-mesenchymal Transition in Breast Cancer and Normal Cells. The phrases "surveyor assay" and "Surveyor Nuclease" have been updated to "T7E1 assay" to " T7 endonuclease I" respectively.
Step 1.2 in the Protocol has been updated from:
- Surveyor nuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by surveyor nuclease assay (also known as T7 endonuclease I (T7EI) assay)17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
to:
- T7 endonuclease assay of sgRNA
NOTE: The targeting efficiency of the sgRNA used for the knock-in experiment is evaluated by T7 endonuclease (T7EI) assay17. Select the sgRNA with high DNA cleavage efficiency and a low distance between the sgRNA cutting site and the stop codon.
Figure 1 in the Representative Results has been updated from:
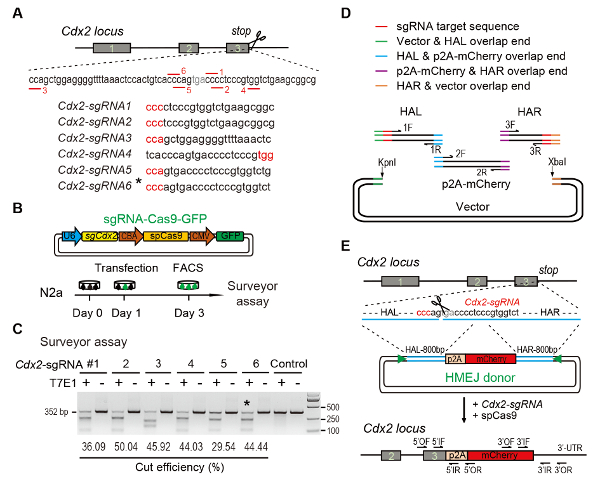
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for surveyor assay. (C) Surveyor assay for Cdx2 targeting: 6 different sgRNAs were designed for surveyor assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
to:
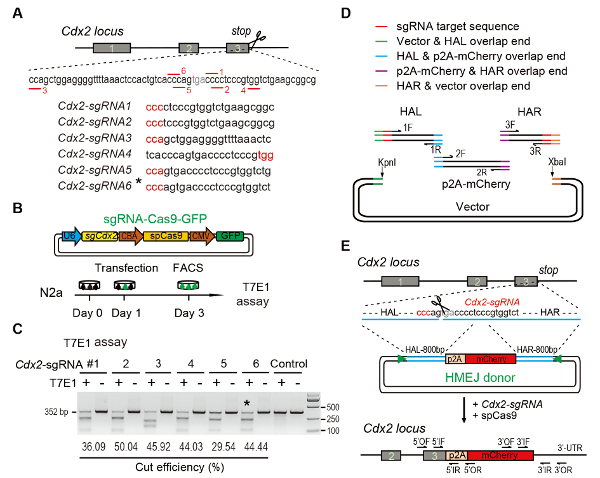
Figure 1: HMEJ-mediated targeted integration in vitro.
(A) Experimental scheme for selection of sgRNAs: Six different sgRNAs (Cdx2-sgRNA1~Cdx2-sgRNA6) around the stop codon of the Cdx2 locus with a higher rank and off-target potential were chosen based on online CRISPR design tool. The protospacer adjacent motif (PAM) sequence is in red. (B) Experimental design: The Cas9-CMV-GFP expression plasmids expressing sgRNA, Cas9, and GFP were introduced into N2a cells. GFP+ cells were sorted at day 3 for T7EI assay. (C) T7EI assay for Cdx2 targeting: 6 different sgRNAs were designed for T7EI assay. Normal N2a cell genomic DNA serves as control. *, the sgRNA used for Cdx2-2A-mCherry knock-in experiment. (D) Schematic overview of construction of HMEJ donors using Gibson assembly. (E) Schematic overview of HMEJ-mediated gene targeting strategy at Cdx2 locus. HAL/HAR, left/right homology arm; triangles, sgRNA target sites; OF/OR, outer forward/reverse primer; IF/IR, inner forward/reverse primer. Figure modified from previous report10. Please click here to view a larger version of this figure.
