Source: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Department of Clinical Sciences Lund, Division of Infection Medicine, Biomedical Center, Lund University, 221 00 Lund, Sweden
Planet Earth is a habitat for millions of bacterial species, each of which has specific characteristics. Identification of bacterial species is widely used in microbial ecology to determine biodiversity of environmental samples and medical microbiology to diagnose infected patients. Bacteria can be classified using conventional microbiology methods, such as microscopy, growth on specific media, biochemical and serological tests, and antibiotic sensitivity assays. In recent decades, molecular microbiology methods have revolutionized bacterial identification. A popular method is 16S ribosomal RNA (rRNA) gene sequencing. This method is not only faster and more accurate than conventional methods, but also allows identification of strains that are difficult to grow in laboratory conditions. Furthermore, differentiation of strains at the molecular level enables discrimination between phenotypically identical bacteria (1-4).
16S rRNA joins with a complex of 19 proteins to form a 30S subunit of the bacterial ribosome (5). It is encoded by the 16S rRNA gene, which is present and highly conserved in all bacteria due to its essential function in ribosome assembly; however, it also contains variable regions which may serve as fingerprints for particular species. These features have made the 16S rRNA gene an ideal genetic fragment to be used in identification, comparison, and phylogenetic classification of bacteria (6).
16S rRNA gene sequencing is based on the polymerase chain reaction (PCR) (7-8) followed by DNA sequencing (9). PCR is a molecular biology method used to amplify specific fragments of DNA through a series of cycles that include:
i) Denaturation of a double stranded DNA template
ii) Annealing of primers (short oligonucleotides) that are complementary to the template
iii) Extension of primers by the DNA polymerase enzyme, which synthesizes a new DNA strand
A schematic overview of the method is shown in Figure 1.
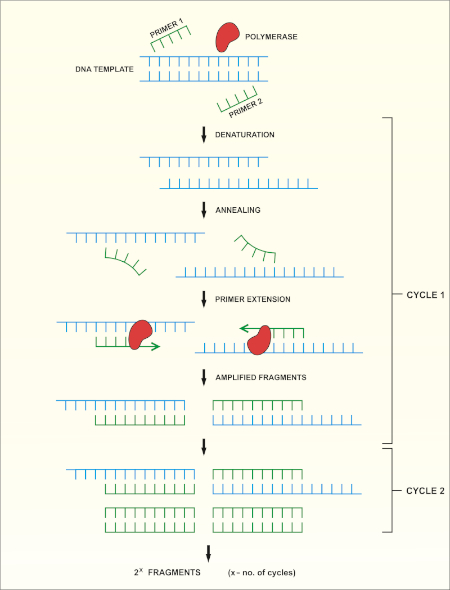
Figure 1: Schematic overview of the PCR reaction. Please click here to view a larger version of this figure.
There are several factors that are important for a successful PCR reaction, one of which is quality of the DNA template. Isolation of chromosomal DNA from bacteria can be performed using standard protocols or commercial kits. Special care should be taken to obtain DNA that is free of contaminants that can inhibit the PCR reaction.
Conserved regions of the 16S rRNA gene permit the design of universal primer pairs (one forward and one reverse) that can bind to and amplify the target region in any bacterial species. The target region can vary in size. While some primer pairs can amplify most of the 16S rRNA gene, others amplify only parts of it. Examples of commonly used primers are shown in Table 1 and their binding sites are depicted in Figure 2.
| Primer name | Sequence (5'→3') | Forward/ reverse | Reference |
| 8F b) | AGAGTTTGATCCTGGCTCAG | forward | -1 |
| 27F | AGAGTTTGATCMTGGCTCAG | forward | -10 |
| 515F | GTGCCAGCMGCCGCGGTAA | forward | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | reverse | -12 |
| 1391R | GACGGGCGGTGTGTRCA | reverse | -11 |
| 1492R | GGTTACCTTGTTACGACTT | reverse | -11 |
Table 1: Examples of standard oligonucleotides used in amplification of 16S rRNA genes a).
a) The expected lengths of the PCR product generated using the different primer combinations can be estimated by calculating the distance between the binding sites for the forward and the reverse primer (see Figure 2), e.g. the size of the PCR product using primer pair 8F-1492R is ~1500 bp, and for primer pair 27F-911R ~900 bp.
b) also known as fD1

Figure 2: Representative figure of the 16S rRNA sequence and the primer-binding sites. Conserved regions are colored in grey and variable regions are filled with diagonal lines. To allow for the highest resolution, primer 8F and 1492R (name based on location on rRNA sequence) are used to amplify the whole sequence, allowing for the sequencing of several variable regions of the gene. Please click here to view a larger version of this figure.
Cycling conditions for PCR (i.e. the temperature and time required for the DNA to be denatured, annealed with primers, and synthesized) are dependent on the type of polymerase that is used and the properties of the primers. It is recommended to follow the manufacturer's guidelines for a particular polymerase.
After the PCR program is completed, the products are analyzed by agarose gel electrophoresis. A successful PCR yields a single band of expected size. The product must be purified prior to sequencing to remove residual primers, deoxyribonucleotides, polymerase, and buffer which were present in the PCR reaction. The purified DNA fragments are usually sent for sequencing to commercial sequencing services; however, some institutions perform DNA sequencing at their own core facilities.
The DNA sequence is automatically generated from a DNA chromatogram by a computer and must be carefully checked for quality, as manual editing is sometimes needed. Following this step, the gene sequence is compared with sequences deposited in the 16S rRNA database. The regions of similarity are identified, and the most similar sequences are delivered.