מקור: אווה בוקובסקה-פאניבנד1, עדדה אנדרסון1, רולף לוד1
1 המחלקה למדעים קליניים לונד, החטיבה לרפואת זיהומים, המרכז הביו-רפואי, אוניברסיטת לונד, 221 00 לונד, שוודיה
כדור הארץ הוא בית גידול למיליוני מינים חיידקיים, שלכל אחד מהם מאפיינים ספציפיים. זיהוי של מינים חיידקיים נמצא בשימוש נרחב באקולוגיה מיקרוביאלית כדי לקבוע את המגוון הביולוגי של דגימות סביבתיות ומיקרוביולוגיה רפואית לאבחון חולים נגועים. חיידקים יכולים להיות מסווגים בשיטות מיקרוביולוגיות קונבנציונליות, כגון מיקרוסקופיה, צמיחה על מדיה ספציפית, בדיקות ביוכימיות וסרולוגיות, ובדיקות רגישות לאנטיביוטיקה. בעשורים האחרונים, שיטות מיקרוביולוגיה מולקולרית חוללו מהפכה בזיהוי חיידקים. שיטה פופולרית היא ריצוף גנים RNA ריבוזומלי (rRNA) 16S. שיטה זו היא לא רק מהירה ומדויקת יותר מאשר שיטות קונבנציונליות, אלא גם מאפשר זיהוי של זנים שקשה לגדול בתנאי מעבדה. יתר על כן, הבחנה של זנים ברמה המולקולרית מאפשרת אפליה בין חיידקים זהים פנוטיפיים (1-4).
16S rRNA מצטרף לתסביך של 19 חלבונים ליצירת תת-unit של ריבוזום חיידקי (5). הוא מקודד על ידי הגן 16S rRNA, אשר קיים ושמובטח מאוד בכל החיידקים בשל תפקידו החיוני בהרכבת ריבוזום; עם זאת, הוא מכיל גם אזורים משתנים אשר עשוי לשמש טביעות אצבעות עבור מינים מסוימים. תכונות אלה הפכו את גן 16S rRNA לשבר גנטי אידיאלי לשימוש בזיהוי, השוואה וסיווג פילוגנטי של חיידקים (6).
ריצוף גנים rRNA 16S מבוסס על תגובת שרשרת פולימראז (PCR) (7-8) ואחריו ריצוף DNA (9). PCR היא שיטת ביולוגיה מולקולרית המשמשת להגברת שברי DNA ספציפיים באמצעות סדרה של מחזורים הכוללים:
i) דנטורציה של תבנית DNA כפולה תקועה
ii) חישול פריימרים (אוליגונוקלאוטידים קצרים) המשלימים לתבנית
iii) הרחבת פריימרים על ידי אנזים פולימראז DNA, אשר סינתזה גדיל DNA חדש
מבט כולל סכמטי על פעולת השירות מוצג באיור 1.
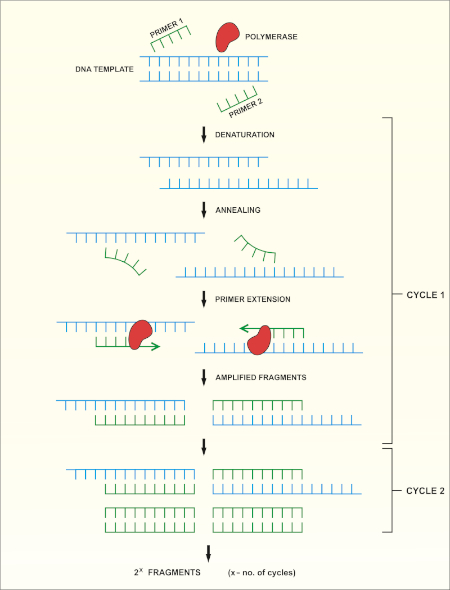
איור 1: מבט כולל סכמטי על תגובת ה- PCR. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
ישנם מספר גורמים החשובים לתגובת PCR מוצלחת, שאחד מהם הוא איכות תבנית ה- DNA. בידוד של DNA כרומוזומלי מחיידקים יכול להתבצע באמצעות פרוטוקולים סטנדרטיים או ערכות מסחריות. יש לנקוט בזהירות מיוחדת כדי להשיג DNA כי הוא ללא מזהמים שיכולים לעכב את תגובת PCR.
אזורים שמורים בגן 16S rRNA מאפשרים תכנון של זוגות פריימר אוניברסליים (אחד קדימה ואחד הפוך) שיכולים להיקשר ולהגביר את אזור היעד בכל מין חיידקי. אזור היעד עשוי להשתנות בגודלו. בעוד שזוגות פריימר מסוימים יכולים להגביר את רוב הגן 16S rRNA, אחרים מגבירים רק חלקים ממנו. דוגמאות של פריימרים נפוצים מוצגות בטבלה 1 ואתרי האיגוד שלהם מתוארים באיור 2.
| שם פריימר | רצף (5’→3′) | קדימה/הפוך | הפניה |
| 8F b) | AGAGTTTGATCCTGGCTCAG | קדימה | -1 |
| 27F | AGAGTTTGATCMTGGCTCAG | קדימה | -10 |
| 515F | GTGCCAGCMGCCGCGGTAA | קדימה | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | הפוך | -12 |
| 1391R | GACGGGCGGTGTGTRCA | הפוך | -11 |
| 1492R | GGTTACCTTGTTACGACTT | הפוך | -11 |
טבלה 1: דוגמאות של אוליגונוקלאוטידים סטנדרטיים המשמשים להגברה של 16S rRNA גנים a).
א) ניתן להעריך את האורכים הצפויים של מוצר ה- PCR שנוצר באמצעות שילובי פריימר שונים על-ידי חישוב המרחק בין אתרי האיגוד עבור הפוריימר קדימה לבין הפריימר ההפוך (ראה איור 2), למשל גודל מוצר ה- PCR באמצעות זוג פריימר 8F-1492R הוא ~ 1500 bp, ולזוג פריימר 27F-911R ~ 900 bp.
ב) הידוע גם בשם fD1

איור 2: דמות מייצגת של רצף rRNA 16S ואתרי איגוד הפריימר. אזורים שמורים נצבעים באפור ואזורים משתנים מלאים בקווים אלכסוניים. כדי לאפשר את הרזולוציה הגבוהה ביותר, פריימר 8F ו- 1492R (שם המבוסס על מיקום על רצף rRNA) משמשים להגברת הרצף כולו, ומאפשרים רצף של מספר אזורים משתנים של הגן. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
תנאי הרכיבה על אופניים עבור PCR (כלומר הטמפרטורה והזמן הנדרשים עבור ה- DNA להיות denatured, חישול עם פריימרים, מסונתז) תלויים בסוג של פולימראז המשמש ואת המאפיינים של פריימרים. מומלץ לעקוב אחר הנחיות היצרן עבור פולימראז מסוים.
לאחר השלמת תוכנית ה- PCR, המוצרים מנותחים על ידי אלקטרופורזה ג’ל אגרוז. PCR מוצלח מניב רצועה אחת בגודל הצפוי. יש לטהר את המוצר לפני הרצף כדי להסיר פריימרים שיורית, deoxyribonucleotides, פולימראז, חוצץ אשר היו נוכחים בתגובת PCR. שברי הדנ”א המטוהרים נשלחים בדרך כלל לרצף לשירותי רצף מסחריים; עם זאת, מוסדות מסוימים מבצעים ריצוף DNA במתקני הליבה שלהם.
רצף הדנ”א נוצר באופן אוטומטי מכרומטוגרמה DNA על ידי מחשב ויש לבדוק אותו בקפידה לאיכות, שכן לעתים יש צורך בעריכה ידנית. לאחר שלב זה, רצף הגנים מושווה לרצפים שהופקדו במסד הנתונים של 16S rRNA. אזורי הדמיון מזוהים, והרצפים הדומים ביותר מועברים.