In this article, we demonstrate the applicability of the TAP method to the well-characterized CycT1-CDK9 complex (also known as P-TEFb kinase).
Plasmids encoding Cyclin T1-STREP (CycT1:S) and CDK9-FLAG (CDK9:F), or CDK9-STREP (CDK9:S) and Cyclin T1-FLAG (CycT1:F) (Table 1), were transfected into HEK293T cells. Negative controls included transfections with an empty vector and the CDK9:F or CycT1:F plasmids (Figures 3A and B, respectively). The proteins were expressed for 48 hr before the cells were collected. Multiple plates of cells were used per experiment (five plates per sample were used for the data presented in Figure 3) to increase protein production and recovery. After the cells were lysed in individual microcentrifuge tubes, the cell lysates of the same sample were combined into one microcentrifuge tube prior to adding the STREP beads. Combining the protein samples with one portion of beads increases the final protein concentration, which can improve protein recovery from proteins that are difficult to express. Following the initial elution, a second elution can be done in order to increase the total protein input for the FLAG IP. It is important to save the input for each purification step before adding the beads for future troubleshooting (Table 2), if needed.
The data in Figure 3A shows the results of the TAP method for CycT1:S and CDK9:F, analyzed by western blot. CDK9:F co-eluted with CycT1:S from the STREP beads, but not in the negative control AP lacking CycT1:S, indicating that CDK9:F and CycT1:S form a protein-protein complex. Expectedly, both proteins were detected in the FLAG elution, which further confirmed the complex formation. Figure 3B shows the results of the reciprocal TAP method, where the epitope tags of the two proteins were swapped (CDK9:S and CycT1:F), and analyzed by western blot.
Figures 3C and 3D show the results of the TAP and reciprocal TAP analyzed by silver staining, respectively. Figure 3C lane 2 showed that CDK9:F co-eluted with CycT1:S off of the STREP beads, but not from the control AP (lane 1), thus indicating that CDK9:F and CycT1:S formed a protein-protein complex. In the subsequent FLAG elution, as shown in lane 4, CycT1:S co-eluted with CDK9:F off of the FLAG beads. Similarly, Figure 3D showed that CycT1:F co-eluted with CDK9:S off of the STREP beads, but not from the control AP, and that the protein-protein complex was efficiently recovered after the second FLAG IP step. Both the western blots and silver stains confirmed that the TAP protocol worked efficiently for the purification, isolation, and characterization of the protein complex of interest.
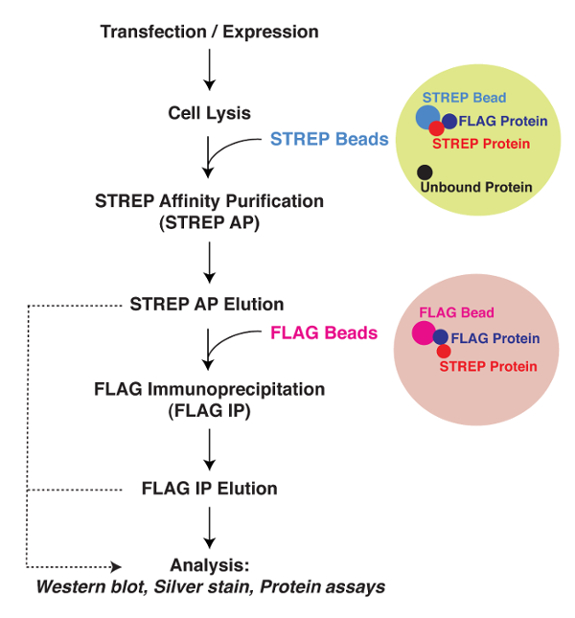
Figure 1: Overview of TAP Purification Strategy. A simple scheme describing the steps of TAP. Plasmids encoding the protein of interest with either the STREP or FLAG epitope tag are co-transfected into mammalian cells. After 48 h induction, the cells are collected and lysed. Soluble cell lysate containing the protein of interest along with other proteins are loaded onto STREP beads for STREP AP. Proteins that either contain the STREP tag or are bound to the STREP-tagged protein attached to the beads are then eluted. The FLAG beads were then added into the STREP elution. Proteins that either contain the FLAG tag or are bound to the FLAG-tagged protein attached to the beads are then eluted. Elutions could be analyzed by the indicated methods. Please click here to view a larger version of this figure.
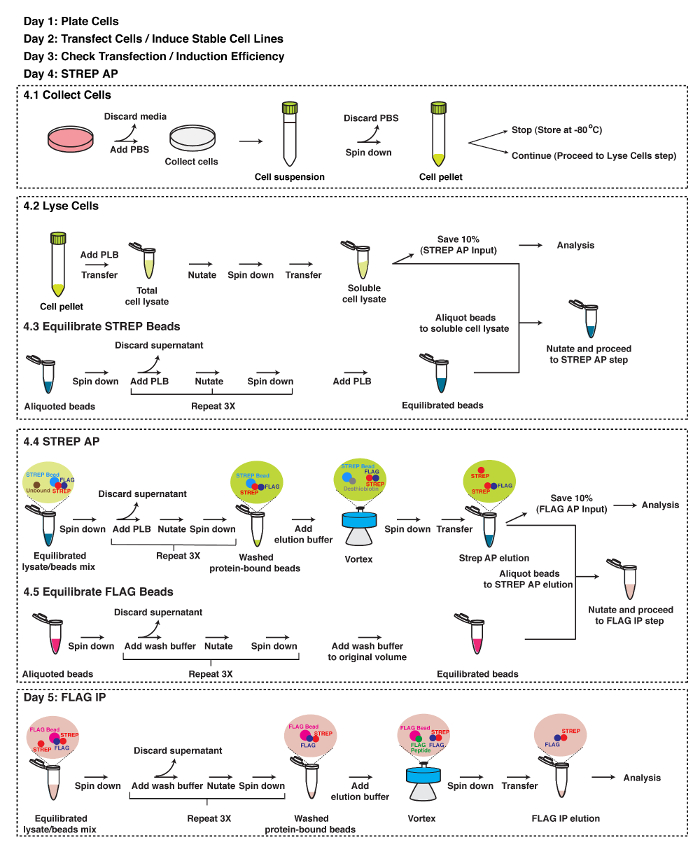
Figure 2: Schematic Representation of TAP Strategy. A detailed protocol of TAP. See text for details. Please click here to view a larger version of this figure.
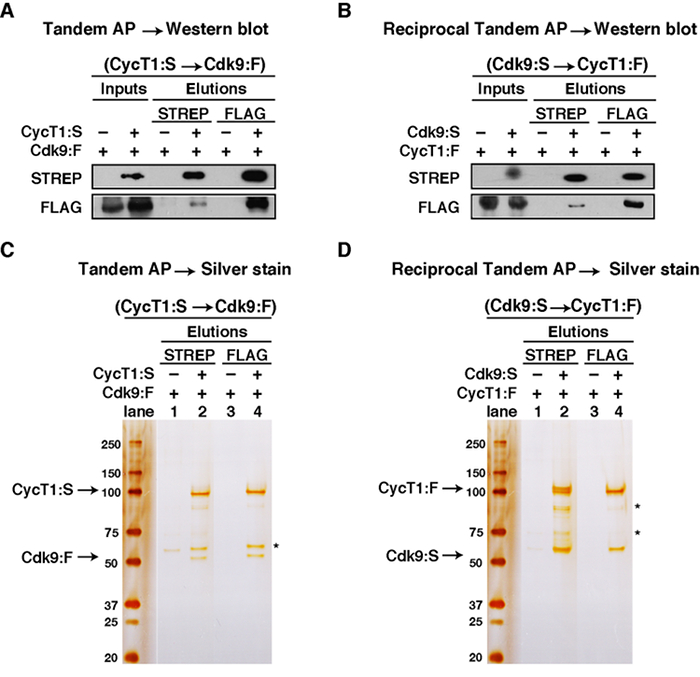
Figure 3: Example of Step-by-step Analysis of TAP Purification. (A) Western blot analysis of TAP (CycT1:S and CDK9:F). (B) Western blot analysis of reciprocal TAP (CDK9:S and CycT1:F). From 1 ml of whole cell lysate, 5 μl (0.2%) was loaded as "STREP AP Input." 975 μl of the remaining whole cell lysate was added to STREP beads for STREP AP and 100 μl of "STREP AP Elution" was collected. From this, 5 μl (2%) was loaded. 80 μl of the "STREP AP Elution" was added to FLAG beads for FLAG IP and 30 μl of "FLAG IP Elution" was collected. From the 30 μl of "FLAG IP Elution", 7 μl (23.3%) was loaded. (C) Silver stain analysis of TAP (CycT1:S and CDK9:F). (D) Silver stain analysis of reciprocal TAP (CDK9:S and CycT1:F). 5 μl (2%) of "STREP AP Elution" and 7 μl (23.3%) of "FLAG IP Elution" were loaded. The asterisks indicate co-eluted impurities. Please click here to view a larger version of this figure.
| Protein | Residues | Vector | Tag | Cloning sites | Reference |
| CycT1 | 1-708 | pcDNA/4TO | STREP | HindIII – EcoRI | McNamara et al., 201310 |
| CycT1 | 1-708 | pcDNA/4TO | FLAG | HindIII – EcoRI | This article |
| CDK9 | 1-372 | pcDNA/4TO | STREP | HindIII – XhoI | This article |
| CDK9 | 1-372 | pcDNA/4TO | FLAG | HindIII – XhoI | McNamara et al., 201310 |
| GFP | 1-238 | pcDNA/4TO | STREP-FLAG | BamHI – XhoI | This article |
Table 1: Plasmids used in this Study. CycT1 and CDK9 were ligated into the pcDNA/4TO-STREP and pcDNA/4TO-FLAG plasmid vectors. These constructs were transformed into DH5α competent cells and plated on LB-Ampicillin plates. Colonies were selected, grown, and screened. Successfully ligated clones were verified by Sanger sequencing and the combination of restriction digestion and agarose gel electrophoresis. For CycT1 we used a shorter version (1 – 708) instead of the full-length (1 – 726) because the last 18 residues contain a PEST sequence (a peptide sequence rich in proline, glutamic acid, serine, and threonine), which acts as a signal peptide for protein degradation20. CDK9 was full-length. The sequence of the tandem STREP tag is as follows: GGGGWSHPQFEKGGGSGGGSGGGSWSHPQFEK. The sequence of the tandem FLAG tag is as follows: GGGGDYKDHDGDYKDHDIDYKDDDDK.
Table 2: Troubleshooting Table. Before beginning with troubleshooting, double check to make sure that each essential step has been followed. Certain steps in this protocol require more optimization than others to achieve the expected result. Please click here to download this file.
