Methylated RNA Immunoprecipitation Assay to Study m5C Modification in Arabidopsis
Summary
The methylated RNA immunoprecipitation assay is an antibody-based method used to enrich for methylated RNA fragments. Coupled with deep sequencing, it leads to the identification of transcripts carrying the m5C modification.
Abstract
Secondary base modifications on RNA, such as m5C, affect the structure and function of the modified RNA molecules. Methylated RNA Immunoprecipitation and sequencing (MeRIP-seq) is a method that aims to enrich for methylated RNA and ultimately identify modified transcripts. Briefly, sonicated RNA is incubated with an antibody for 5-methylated cytosines and precipitated with the assistance of protein G beads. The enriched fragments are then sequenced and the potential methylation sites are mapped based on the distribution of the reads and peak detection. MeRIP can be applied to any organism, as it does not require any prior sequence or modifying enzyme knowledge. In addition, besides fragmentation, RNA is not subjected to any other chemical or temperature treatment. However, MeRIP-seq does not provide single-nucleotide prediction of the methylation site as other methods do, although the methylated area can be narrowed down to a few nucleotides. The use of different modification-specific antibodies allows MeRIP to be adjusted for the different base modifications present on RNA, expanding the possible applications of this method.
Introduction
In all three kingdoms of life, RNA species undergo post-transcriptional modifications and research on these functionally relevant biochemical modifications is called “epitranscriptomics”. Epitranscriptomics is a growing field and various methods are being developed to study and map the modifications on RNA molecules (reviewed in1,2). More than a hundred RNA modifications have been found, detected in rRNAs, tRNAs, other ncRNAs, as well as mRNAs3,4. Although the presence and function of chemically diverse post-transcriptional modifications in tRNAs and rRNAs are extensively studied5,6,7,8, only recently have mRNA modifications been characterized. In plants, many mRNA modifications have been identified to date, including m7G at the cap structure9, m1A10, hm5C11,12, and uridylation13. However, only m6A10,14,15, m5C11,16,17, and pseudouridine18 have been mapped transcriptome-wide in Arabidopsis. Post-transcriptional mRNA base modifications are involved in several developmental processes19,20.
One of the most commonly used approaches in epitranscriptomics is the methylated RNA immunoprecipitation coupled with deep sequencing (MeRIP-seq). MeRIP-seq was developed in 2012 to study m6A in mammalian cells21,22. It requires the use of an antibody for the desired modification and aims to enrich for RNA fragments carrying the modified nucleotide(s). It is usually followed by deep sequencing to identify and map the enriched fragments or quantitative PCR to verify specific RNA targets. The accuracy of MeRIP is based on the specificity of the antibody to recognise the modified nucleotide over similar modifications (e.g., m5C and hm5C11,23). Besides m6A, MeRIP-seq has been also applied to study m1A and m5C RNA methylation in several organisms11,17,23,24,25.
Methylation of the cytosine at the fifth carbon position (m5C) is the most prevalent DNA modification26,27 and one of the most common RNA modifications too3,4. While m5C was detected in eukaryotic mRNAs in 197528, only recently have studies focused on mapping the modification transcriptome-wide, in coding and non-coding RNAs11,16,17,23,29,30,31,32,33,34.
Alternative methods used in m5C RNA research include chemical conversion of non-methylated cytosines into uracils (bisulfite sequencing) and immunoprecipitation assays based on an irreversible binding of a known RNA cytosine methyltransferase to its RNA targets (miCLIP, aza-IP). In brief, bisulfite sequencing exploits the feature of 5-methylated cytosine to be resistant to sodium bisulfite treatment that deaminates unmodified cytosines to uracil. The method was first developed for DNA but adapted for RNA too and many studies have chosen this approach to detect m5C sites in RNA16,23,29,32,34,35. Both miCLIP and aza-IP require previous knowledge of the RNA cytosine methyltransferase and use of the respective antibody. In the case of miCLIP (methylation individual-nucleotide-resolution crosslinking and immunoprecipitation), the methyltransferase carries a single amino acid mutation so that it binds to the RNA substrate but cannot be released30. In aza-IP (5-azacytidine–mediated RNA immunoprecipitation), the irreversible binding is formed between the 5-azaC nucleoside and the RNA cytosine methyltransferase when exogenously provided 5-azaC is incorporated by RNA polymerases into a target RNA molecule31.
The main advantage of these three methods is that they allow single nucleotide resolution mapping of m5C. In addition, miCLIP and aza-IP provide information about the specific targets of a selected RNA cytosine methyltransferase, deciphering deeper the mechanism and role of post-transcriptional RNA modifications. However, the MeRIP-seq approach can identify transcriptome-wide m5C regions without any previous knowledge required and avoids harsh chemical and temperature conditions, such as bisulfite treatment or incubation with 5-azaC. Both MeRIP and bisulfite sequencing can be inhibited by secondary RNA structures36. The fragmentation step that is included in the MeRIP assay prior to immunoprecipitation aims to facilitate antibody binding and increase the resolution of m5C identification.
Another method worth mentioning is mass spectrometry (MS) of RNA nucleosides. MS can detect and distinguish any type of modification both on DNA and on RNA. Briefly, RNA is extracted and DNase digested, then desalted and digested to single nucleosides. The RNA nucleosides are analyzed by a mass spectrometer. This method can be used to quantify the levels of each modification and it does not rely on an antibody or a chemical conversion. However, a major drawback is that it provides bulk information about the presence of RNA modifications. In order to map the modifications, MS needs to be combined with RNase digestion and sequencing information about specific RNA molecules, as in the case of human tRNALeu(CAA)37.
Here, we describe and discuss the MeRIP assay as used to study m5C RNA methylation in Arabidopsis17.
Protocol
1. Preparing the RNA
- Grind 200 mg of plant tissue to powder in liquid nitrogen, making sure that the tissue remains frozen throughout the procedure.
- Extract the RNA from the desired plant tissue following an acid guanidinium thiocyanate-phenol-chloroform extraction protocol. To decrease the possibility to contaminate RNA with DNA during the phase separation, use 1-bromo-3-chloropropane instead of chloroform.
- Add 1 mL of RNA extraction reagent containing guanidine thiocyanate and acid phenol to the grinded plant tissue (500 µL per 100 mg tissue). Mix well by inverting and make sure all the tissue is wet. Incubate for 10 min at room temperature to dissociate the ribonucleoprotein complexes.
- Centrifuge for 10 min at 12,000 x g at 4 °C and transfer the supernatant to a new 1.5 mL tube.
- Add 200 µL of 1-bromo-3-chloropropane (100 µL per 500 µL RNA extraction reagent) and vortex vigorously.
- Centrifuge for 15 min at 12,000 x g at 4 °C and transfer the upper aqueous phase (approx. 500 µL) to a new 1.5 mL tube.
- Add 1 volume of isopropanol (500 µL) and 0.1 volume of 3 M sodium acetate pH 5.5 (50 µL), mix well by inverting and precipitate 10 min at -20 °C.
NOTE: The use of sodium acetate (NaOAc) is recommended in order to enhance RNA precipitation. The protocol can be paused at this point by prolonging the precipitation of RNA for a few hours or even overnight. - Centrifuge for 30 min at 12,000 x g at 4 °C and discard the supernatant.
- Wash the pellet twice with 500 µL of 80% EtOH, centrifuge for 5 min at 12,000 x g at 4 °C and discard.
- Wash the pellet once with 500 µL 99% EtOH, centrifuge for 5 min at 12,000 x g at 4 °C and discard.
- Dry the pellet for 5-10 min and dissolve in 30 µL of RNase-free H2O.
NOTE: Instead, use any RNA extraction protocol of choice (e.g., a column-based system). If a DNase digestion is included in the protocol, skip it in the following step (step 1.3).
- Measure the RNA concentration (e.g., with the use of a spectrophotometer) and digest 20 µg of RNA with DNase.
NOTE: DNA is rich in m5C and the antibody does not distinguish between DNA and RNA.- In a typical DNase reaction, treat 10 µg of RNA in a 50 µL reaction. Mix the following components and incubate the reaction(s) at 37 °C for 30 min:
10 µg of RNA x µL
10x DNase buffer 5 µL
DNase 1 µL (2 units)
RNase-free H2O up to 50 µL - Remove the enzyme either by adding an appropriate volume of DNase inactivation reagent (if it is included in the DNase kit and according to manufacturer’s instructions) or by performing a cleanup step (e.g., column purification or phenol/chloroform extraction).
- In a typical DNase reaction, treat 10 µg of RNA in a 50 µL reaction. Mix the following components and incubate the reaction(s) at 37 °C for 30 min:
- Check the quality and purity of the isolated RNA by capillary electrophoresis and proceed if the RNA integrity number (RIN) is higher than 7, to ensure the samples are of good quality.
- OPTIONAL: Remove the ribosomal RNA to enrich the samples in mRNA content using an rRNA removal kit and according to manufacturer’s protocol.
- Use the DNase treated RNA from the previous step for the rRNA depletion reaction(s). Perform multiple reactions if the amount of total RNA is more than the maximum amount suggested for the reaction.
- Note that only 5-10% of the input amount will be recovered after rRNA depletion. Proceed with the rRNA depleted RNA (equal amount for all samples) and ignore the amounts mentioned in the following steps, as they refer to total RNA.
NOTE: For a comparison and description of available rRNA depletion methods see references 38,39,40. rRNA is the major part of total RNA and is m5C methylated in many organisms.
- Prepare in advance in vitro transcripts (IVT) to be used as control RNA sequences and add them in the samples.
- Produce two distinct IVTs using an in vitro transcription kit, one with non-methylated nucleosides and one where rCTP is replaced by 5-methyl-rCTP, to serve as negative and positive controls in MeRIP, respectively. The transcripts prepared were those of EGFP and Renilla luciferase.
NOTE: The IVTs should not exist in the transcriptome of the organism you are analysing. If the IVTs are from the same template (e.g., both EGFP), then add the positive and negative control in two different samples. If their sequence is different (e.g., EGFP and Renilla), they can be added to the same sample. - Spike in each sample 0.1 ng IVT per 3 µg of RNA, as controls.
- Produce two distinct IVTs using an in vitro transcription kit, one with non-methylated nucleosides and one where rCTP is replaced by 5-methyl-rCTP, to serve as negative and positive controls in MeRIP, respectively. The transcripts prepared were those of EGFP and Renilla luciferase.
- Sonicate the RNA to approximately 100 nt fragments.
NOTE: The conditions for RNA shearing must be adjusted in advance and they differ for each sonicator. For the model used here, sonication is performed with the following conditions: Peak power 174, Duty factor 10, Cycles/burst 200, 17 min.- Sonicate the same amount of RNA for all samples, at least 12 µg RNA per sample in 80 µL of total volume (min 60 µL, max 100 µL), filled up with RNase-free H2O.
- Confirm the efficiency of sonication and the concentration of the RNA samples by capillary electrophoresis. The average size of fragmented RNA should be around 100 nt.
2. Methylated RNA Immunoprecipitation (MeRIP)
- In low-binding tubes, add 9 µg of sonicated RNA and RNase-free H2O up to 60 µL (or more, depending on the concentration).
- Dissociate the secondary structures by heating the RNA at 70 °C in a water bath for 10 min and cooling down for an additional 10 min in an ice-water mix.
- Split the sample in three parts: one-third (20 µL, if 60 µL were taken in step 2.1) is saved in a separate tube at -80 °C as the Input sample. Fill the remaining 40 µL with RNase-free H2O up to 860 µL and then split in two low-binding tubes: one for IP and one for the Mock control (430 µL each).
- Add to both tubes:
50 µL of 10x MeRIP buffer
10 µL of RNase inhibitor
10 µL of α-m5C antibody (10 µg) in the IP sample per 10 µL of H2O in the Mock sample
NOTE: The antibody clone used previously is not commercially available anymore. However, any anti-5-methylcytosine monoclonal antibody should work similarly. Antibodies should be tested for specificity before used for MeRIP11,23. - Seal the tubes with parafilm and incubate for 12-14 hours at 4 °C, with overhead rotation.
- The next day, prepare the protein G magnetic beads for binding.
- For each tube (either IP or Mock control), use 40 µL of beads. Add the total amount of beads (# of tubes x 40 µL, e.g., for 2 IP and 2 Mock samples, 160 µL of beads are needed) in a 15 mL tube and wash three times with 800 µL of 1x MeRIP buffer per sample (# of tubes x 800 µL buffer, e.g., 3.2 mL for 2 IP and 2 Mock samples).
- Perform washes at room temperature for 5 min with overhead rotation, collect the beads with the help of a magnetic rack and discard the washing buffer. After the third wash, resuspend the beads in the same volume of 1x MeRIP buffer as the initial volume of beads taken (# of tubes x 40 µL, e.g., 160 µL of 1x MeRIP buffer for 2 IP and 2 Mock samples).
NOTE: The amount of protein G beads used is determined by the binding capacity of the beads for the specific antibody type and the amount of antibody used. In this case, the beads have a binding capacity of approx. 8 µg of mouse IgG per mg of beads and 30 mg/mL concentration. Therefore, 40 µL are enough to bind approx. 9.6 µg of antibody.
- Add 40 µL of resuspended beads to each IP and Mock sample and incubate for additional 2 hours at 4 °C, with overhead rotation.
- Place the tubes on a magnetic rack for 1 min and discard the supernatant or save it as a control (non-bound RNA sample).
- Wash the beads 5 times by resuspending in 700 µL of 1x MeRIP buffer supplied with 0.01% Tween 20 and incubating for 10 min at room temperature with overhead rotation.
- Resuspend the washed beads in 200 µL of Proteinase K digestion buffer and add 3.5 µL of Proteinase K. Incubate for 3 hours at 50 °C, shaking at 800 rpm. Occasionally, flick manually the bottom of the tube if a sediment of beads is forming during the incubation.
- Extract the RNA by addition of 800 µL of RNA extraction reagent and following an acid guanidinium thiocyanate-phenol-chloroform extraction protocol, and continue as in step 1.2. To increase visibility of the RNA pellet, a colored co-precipitant can be added in isopropanol at the precipitation step. Resuspend the pellet in 20 µL of RNase-free H2O (or equal to Input volume kept in step 2.3).
3. Downstream analysis
- Submit the Input and IP samples for single end sequencing with 50 bases read length (SE50).
- Trim 3’ end adaptors using cutadapt41 and discard reads that are shorter than 48 nt.
- Map trimmed reads to the Arabidopsis genome (TAIR10 annotation) using STAR42 with a cutoff of 6% for mismatches and maximum intron size of 10 kb. Keep uniquely mapped reads for further analysis.
- Identify enriched RNA fragments in IP samples compared to Input using two distinct methods and consider those that are found significantly enriched by both.
- First, detect MeRIP-seq peaks using MACS2 peak caller43 on pooled IPs versus Input.
- Secondly, follow the analysis for MeRIP-seq peak calling as described in Meyer et al.22 and Yang et al.17.
- Using custom R scripts, divide the genome in distinct 25 nt windows and count the number of uniquely mapped reads for each window based on the position of the last mapped nucleotide (since the reads originate from 100 nt RNA fragments).
- Calculate significantly enriched windows in IP samples compared to Input with the Fisher Exact Test. Use the Benjamini-Hochberg procedure to correct for multiple testing.
- Keep the significantly enriched peaks that span over at least two consecutive windows and discard peaks that cover only one window.
- Identify annotated regions of the genome (transcripts) with significantly enriched peaks found by both methods.
- Alternatively or complementarily, test specific RNA targets for their enrichment in the IP samples.
- Reverse transcribe the RNA (same volume of Input, IP and Mock samples) with random hexamers.
- Perform quantitative real-time PCR on the chosen targets, comparing Input, IP and Mock via the ΔΔCt method.
NOTE: The generated product should not be longer than 100 bp, as this is the average fragmentation size.
Representative Results
A schematic of the method is provided in Figure 1. The first critical steps of the protocol are to obtain RNA of good quality (RIN ≥ 7) and sonicate it to approximately 100 nt fragments. The efficiency of both steps is examined by a chip-based capillary electrophoresis machine. In Figure 2A, a representative run of a good RNA sample is shown. The sample is diluted 1:10 before loading on the chip in order to have a concentration that is in the range of detection of the kit used (5-500 ng/µL). The same sample is also run after sonication and is shown in Figure 2B. Notice the presence of one uniform peak shifted to the left of the diagram, at a size of around 100 nucleotides. The lower concentration is caused both by loss of RNA during fragmentation but also because of the increased volume of the samples (60-100 µL, step 1.7.1).
The quality of the IP and Mock samples can be evaluated by qRT-PCR. To this end, the spiked-in IVTs serve as positive and negative controls: the methylated IVT, where in all cytosine positions there is m5C, is expected to be highly enriched in the IP sample; on the contrary, the non-methylated IVT should not have a difference between IP and Mock. The primers used in the qRT-PCR assay for the two control IVTs (EGFP and Renilla luciferase) are listed in Table 1. Indeed, as shown in Figure 3, around 80% of the methylated IVT was recovered in the IP sample, and only approximately 2% in the Mock. For the non-methylated control, the recovery was below 1% in both IP and Mock samples. This verifies the efficiency of MeRIP that methylated RNA fragments were precipitated and enriched, and is a good indicator that the samples can be used for downstream analysis. In addition, the fold enrichment of the non-methylated IVT (IP to Mock ratio) can be applied as a threshold to estimate significance of enrichment in the qRT-PCR assays.
After aligning the reads to the genome (Figure 4), the peak calling algorithms described in steps 3.4.1 and 3.4.2 are applied to identify the statistically significant windows, enriched in the IP samples compared to the Input. The sequences that correspond to these windows can be used further, for example to search for conserved methylation-related motifs11,17.
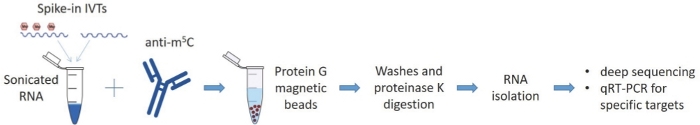
Figure 1: Schematic representation of MeRIP-seq protocol.
RNA samples are incubated with an antibody for 5-methylated cytosines and the complexes are pulled down with protein G magnetic beads that capture the antibodies along with the bound RNA. The eluted RNA samples are analyzed by deep sequencing and qRT-PCR. Please click here to view a larger version of this figure.
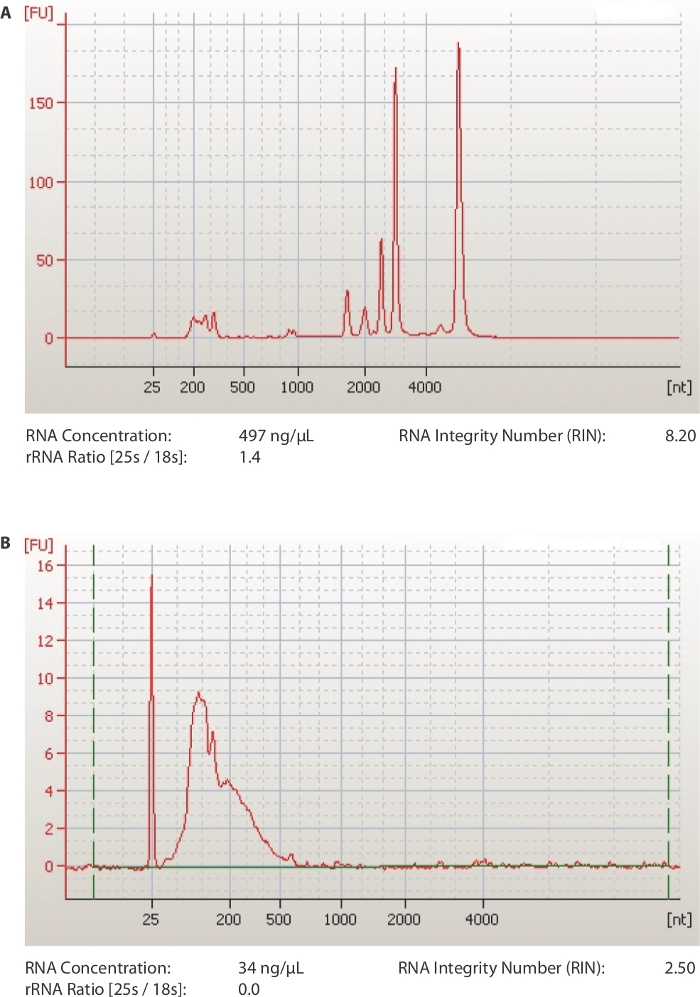
Figure 2: Representative results from quality analysis of RNA samples.
(A) Representative profile of a qualified total RNA plant sample. (B) Representative profile of an RNA sample after sonication to 100 nt fragments. Output files from capillary electrophoresis software. Please click here to view a larger version of this figure.
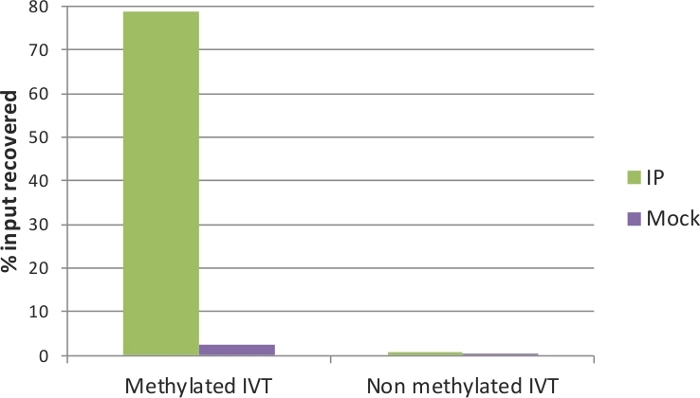
Figure 3: qRT-PCR analysis of control IVTs.
Methylated and non-methylated in vitro transcripts were used as positive and negative controls of the MeRIP assay, respectively. After immunoprecipitation, the methylated IVT is highly enriched in the IP sample (green) but not in the Mock sample (without anti-m5C antibody; purple). The non-methylated IVT showed no enrichment and no difference between IP and Mock. Please click here to view a larger version of this figure.
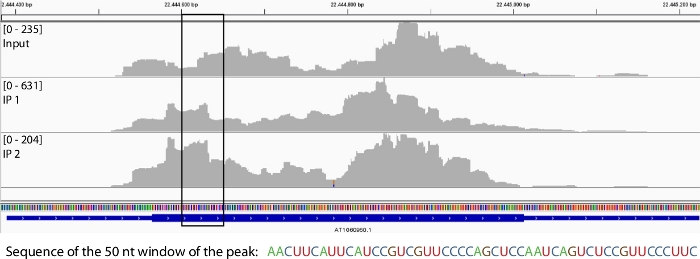
Figure 4: Read alignment before and after MeRIP around a representative transcript.
Reads aligned to a specific transcript in the Input sample (top row) and in two IP replicates (middle and bottom row). The black box shows an identified enriched 50 nt long window based on MACS243 and MeRIP-seq22 peak-calling analyses. Please click here to view a larger version of this figure.
| Target | Primer pair | Product sequence | Product length | ||
| EGFP | For: 5'- GGCAACTACAAGACCCGCGCC -3' | GGCAACTACAAGACCCGCGCCGAG GTGAAGTTCGAGGGCGACACCCTG GTGAACCGCATCGAGCTGAAGGGC |
72 bp | ||
| Rev: 5'- GCCCTTCAGCTCGATGCGGTT -3' | |||||
| Renilla luciferase | For: 5'- GGAGAATAACTTCTTCGTGGAAAC -3' | GGAGAATAACTTCTTCGTGGAAACC ATGTTGCCATCAAAAATCATGAGAA AGTTAGAACCAGAAGAATTTGCAGC |
75 bp | ||
| Rev: 5'- GCTGCAAATTCTTCTGGTTCTAA -3' | |||||
Table 1: Information about the primers and generated products for the qRT-PCR analysis.
Buffer Recipes. Please click here to download this file.
Discussion
RNA carries more than one hundred distinct base modifications4 that form the epitranscriptome44. These modifications add an additional layer of regulation of translation and signalling (reviewed in5,6,8,20,45,46). Early studies were able to detect the presence of post-transcriptional modifications on RNA28,47 but the specific modified RNAs need to be identified in order to understand the role of epitranscriptomics. MeRIP was designed as a method to map RNA methylation sites transcriptome-wide21,22. It can be adapted to any modification, if a specific antibody is available.
The main strength of this protocol is that is relatively simple, safe for the RNA and the user (e.g., 5-azaC is highly toxic for plants and humans) and does not require sequence or modifying enzyme information. Moreover, the enrichment of methylated RNAs by the IP increases the chances of low abundant mRNAs to be detected, unlike bisulfite sequencing that does not contain an enrichment step. When two serial rounds of MeRIP are performed, enrichment in RNA fragments containing methylation sites increases further22. One of the limitations of MeRIP, especially when applied to mRNA methylation studies, is the high quantity of RNA required as input for the assay. The ribodepletion – or poly(A) enrichment – step will reduce the background caused by the heavily modified ribosomal RNA but it removes more than 90% of total RNA. DNA must also be completely removed as it is rich in 5-methylated cytosines. Another drawback is the lower resolution of the exact position of methylation. Sonication of the RNA prior to incubation with the antibody helps towards this direction by narrowing down the region containing the modification to 100-200 nucleotides. When MeRIP is combined with deep sequencing, the resolution of m5C site prediction increases as the sequencing reads form a Gaussian distribution around the potential methylation site. Additionally, the specificity of the antibody needs to be confirmed prior to the assay (e.g., with RNA dot blot assays, performed with oligos synthesized with modified nucleotides), however, to what extent an antibody can actually distinguish between closely related modifications (e.g., m6A and m6Am) is a point of argument in the field48,49. Moreover, highly structured RNAs might interfere with the antibody–antigen interaction, another restriction that is mostly addressed with fragmentation and denaturation of RNA prior to IP. On the contrary, bisulfite sequencing that is also affected by secondary structures, does not include a fragmentation step and this might be one reason that causes discrepancy between the m5C sites and mRNAs predicted by bisulfite sequencing16 and MeRIP-seq11,17. Other cytosine modifications (e.g., hm5C) are also resistant to bisulfite-mediated deamination35.
Modifications of MeRIP-seq include a crosslinking step, either with the introduction of a photoactivatable ribonucleoside (photo-crosslinking-assisted m6A-seq, PA-m6A-seq 50) or using UV light to create antibody-RNA crosslinks after the IP (miCLIP49, different method than the miCLIP described in introduction30, but also with individual-nucleotide resolution). In the future, and as knowledge on RNA methylation is accumulating, more targeted approaches might be preferable, based on the modifying enzymes and/or the consensus sequences where methylation is appearing. The identification of reader proteins is essential to the understanding of the molecular and signalling function of post-transcriptional modifications. Nanopore sequencing technology already allows the direct identification of modified nucleotides without prior treatment of the RNA17 but there is still room for improvement on this field regarding sequence depth and bioinformatic analysis. Overall, MeRIP-seq is currently an established, reliable, and unbiased approach to identify methylated RNA transcripts.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by an IMPRS PhD stipend to E.S, an EMBO Long-Term Fellowship to V.P., and MPI-MPP internal funds to F.K. Part of this work was also funded through PLAMORF, a project that has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant agreement No. 810131). The authors would like to thank Federico Apelt for the bioinformatic analysis and comments on the manuscript, and Mathieu Bahin and Amira Kramdi for the bioinformatic analysis. Work in the Colot lab is supported by Investissements d’Avenir ANR-10-LABX-54 MEMO LIFE, 506 ANR-11-IDEX-0001-02 PSL* Research University.
Materials
| α‐m5C antibody | ZymoResearch | A3001 (discontinued), A3002 available | Clone 10G4 (discontinued), 7D21 available |
| Chip and reagents for capillary electrophoresis | Agilent | 5067-1511 | RNA 6000 Nano kit |
| Dnase | Ambion | AM1907 | TURBO DNA-free kit |
| Co-precipitant of nucleic acids | Ambion | AM9515 | Glycoblue, 15 mg/mL |
| In vitro transcription kit | Promega | P1300 | T7 RiboMAX Large Scale RNA Production System |
| Low-binding reaction tubes | Kisker Biotech | G017 | 1.7 ml microcentrifuge tubes |
| Protein G magnetic beads | Invitrogen | 10003D | Dynabeads Protein G |
| Proteinase K | Ambion | AM2546 | 20mg/mL stock solution, RNA grade |
| RNase inhibitor | Promega | N2615 | RNasin Plus 40 U/μl |
| rRNA removal kit | Epicentre | RZPL11016 | Ribo-Zero rRNA removal kit (Plant leaf) |
| Sonication tubes | Covaris | 520045 | microtube AFA Fiber Pre-Slit Snap-Cap 6x16mm |
| RNA extraction reagent | Ambion | 15596018 | TRIzol reagent |
| Equipment | |||
| Capillary electrophoresis machine | Agilent | G2939BA | 2100 Bioanalyzer System |
| Magnetic rack | Invitrogen | 12321D | DynaMag-2 |
| Microentrifuge | Eppendorf | discontinued | 5417R model |
| Rotator | Benchmark | R4040 | RotoBot Mini |
| Sonicator | Covaris | 500217 | S220 Focused-ultrasonicator |
| Spectrophotometer | ThermoFisher Scientific | ND-ONEC-W | NanoDrop OneC |
| Waterbath | GFL | 1012 | 1012 Incubation bath |
References
- Trixl, L., Lusser, A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdisciplinary Reviews: RNA. 10 (1), 1-17 (2019).
- Mongan, N. P., Emes, R. D., Archer, N. Detection and analysis of RNA methylation. F1000Research. 8, 559 (2019).
- Cantara, W. A., et al. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Research. 39, 195-201 (2011).
- Boccaletto, P., et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Research. 46 (1), 303-307 (2018).
- Agris, P. F. Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO reports. 9 (7), 629-635 (2008).
- Chow, C. S., Lamichhane, T. N., Mahto, S. K. Expanding the Nucleotide Repertoire of the Ribosome with Post-Transcriptional Modifications. ACS Chemical Biology. 2 (9), 610-619 (2007).
- Gigova, A., Duggimpudi, S., Pollex, T., Schaefer, M., Koš, M. A cluster of methylations in the domain IV of 25S rRNA is required for ribosome stability. RNA. 20 (10), 1632-1644 (2014).
- Motorin, Y., Helm, M. tRNA Stabilization by Modified Nucleotides. 생화학. 49 (24), 4934-4944 (2010).
- Shatkin, A. Capping of eucaryotic mRNAs. Cell. 9 (4), 645-653 (1976).
- Shen, L., et al. N 6 -Methyladenosine RNA Modification Regulates Shoot Stem Cell Fate in Arabidopsis. Developmental Cell. 38 (2), 186-200 (2016).
- Cui, X., et al. 5-Methylcytosine RNA Methylation in Arabidopsis Thaliana. Molecular Plant. 10 (11), 1387-1399 (2017).
- Huber, S. M., et al. Formation and abundance of 5-hydroxymethylcytosine in RNA. ChemBioChem. 16 (5), 752-755 (2015).
- Zuber, H., et al. Uridylation and PABP Cooperate to Repair mRNA Deadenylated Ends in Arabidopsis. Cell Reports. 14 (11), 2707-2717 (2016).
- Luo, G. Z., et al. Unique features of the m6A methylome in Arabidopsis thaliana. Nature Communications. 5 (1), 5630 (2014).
- Wan, Y., et al. Transcriptome-wide high-throughput deep m6A-seq reveals unique differential m6A methylation patterns between three organs in Arabidopsis thaliana. Genome Biology. 16 (1), 1-26 (2015).
- David, R., et al. Transcriptome-Wide Mapping of RNA 5-Methylcytosine in Arabidopsis mRNAs and Noncoding RNAs. The Plant Cell. 29 (3), 445-460 (2017).
- Yang, L., et al. m5C Methylation Guides Systemic Transport of Messenger RNA over Graft Junctions in Plants. Current Biology. 29 (15), 2465-2476 (2019).
- Sun, L., et al. Transcriptome-wide analysis of pseudouridylation of mRNA and non-coding RNAs in Arabidopsis. Journal of Experimental Botany. 70 (19), 5089 (2019).
- Chmielowska-Bąk, J., Arasimowicz-Jelonek, M., Deckert, J. In search of the mRNA modification landscape in plants. BMC Plant Biology. 19 (1), 421 (2019).
- Liang, Z., Riaz, A., Chachar, S., Ding, Y., Du, H., Gu, X. Epigenetic Modifications of mRNA and DNA in Plants. Molecular Plant. 13 (1), 14-30 (2020).
- Dominissini, D., et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 485 (7397), 201-206 (2012).
- Meyer, K. D., et al. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell. 149 (7), 1635-1646 (2012).
- Edelheit, S., Schwartz, S., Mumbach, M. R., Wurtzel, O., Sorek, R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS genetics. 9 (6), 1003602 (2013).
- Dominissini, D., et al. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature. 530 (7591), 441-446 (2016).
- Li, X., et al. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nature Chemical Biology. 12 (5), 311-316 (2016).
- Hotchkiss, R. D. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. Journal of Biological Chemistry. 175 (175), 315-332 (1948).
- Wyatt, G. R. Occurence of 5-Methyl-Cytosine in Nucleic Acids. Nature. 166, 237-238 (1950).
- Dubin, D. T., Taylor, R. H. The methylation state of poly A-containing- messenger RNA from cultured hamster cells. Nucleic Acids Research. 2 (10), 1653-1668 (1975).
- Amort, T., et al. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biology. 18 (1), 1-16 (2017).
- Hussain, S., et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Reports. 4 (2), 255-261 (2013).
- Khoddami, V., Cairns, B. R. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nature biotechnology. 31 (5), 458-464 (2013).
- Squires, J. E., et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Research. 40 (11), 5023-5033 (2012).
- Yang, X., et al. 5-methylcytosine promotes mRNA export – NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Research. 27 (5), 606-625 (2017).
- Burgess, A., David, R., Searle, I. R. Conservation of tRNA and rRNA 5-methylcytosine in the kingdom Plantae. BMC Plant Biology. 15 (1), 199 (2015).
- Schaefer, M., Pollex, T., Hanna, K., Lyko, F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Research. 37 (2), (2009).
- Weber, M., et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature Genetics. 37 (8), 853-862 (2005).
- Auxilien, S., Guérineau, V., Szweykowska-Kulińska, Z., Golinelli-Pimpaneau, B. The human tRNA m (5) C methyltransferase Misu is multisite-specific. RNA Biology. 9 (5), 1331-1338 (2012).
- Adiconis, X., et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nature Methods. 10 (7), 623-629 (2013).
- Petrova, O. E., Garcia-Alcalde, F., Zampaloni, C., Sauer, K. Comparative evaluation of rRNA depletion procedures for the improved analysis of bacterial biofilm and mixed pathogen culture transcriptomes. Scientific Reports. 7 (1), 41114 (2017).
- Huang, Y., Sheth, R. U., Kaufman, A., Wang, H. H. Scalable and cost-effective ribonuclease-based rRNA depletion for transcriptomics. Nucleic Acids Research. 48 (4), 20 (2020).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 17 (1), 10 (2011).
- Dobin, A., et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29 (1), 15-21 (2013).
- Zhang, Y., et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biology. 9 (9), 137 (2008).
- Saletore, Y., et al. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biology. 13 (10), 175 (2012).
- Schwartz, S. Cracking the epitranscriptome. RNA. 22 (2), 169-174 (2016).
- Zhao, B. S., Roundtree, I. A., He, C. Post-transcriptional gene regulation by mRNA modifications. Nature Reviews Molecular Cell Biology. 18 (1), 31-42 (2016).
- Keith, G. Mobilities of modified ribonucleotides on two-dimensional cellulose thin-layer chromatography. Biochimie. 77 (1-2), 142-144 (1995).
- Feederle, R., Schepers, A. Antibodies specific for nucleic acid modifications. RNA Biology. 14 (9), 1089-1098 (2017).
- Linder, B., et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nature Methods. 12 (8), 767-772 (2015).
- Chen, K., et al. High-Resolution N 6 -Methyladenosine (m 6 A) Map Using Photo-Crosslinking-Assisted m 6 A Sequencing. Angewandte Chemie International Edition. 54 (5), 1587-1590 (2015).
