1. Preparation
Before starting, assemble the required surgical instruments, dissection and culture media, and 7 day old rat or mouse pups. The protocol is the same for mouse and rat preparations.
(i) Materials and equipment
- Operating scissors (straight length 6″, cat# RS6818, Roboz, Gaithersburg, MD)
- Micro dissecting scissors, (length 4′”, cat# RS5910. Roboz, Gaithersburg, MD)
- Carbon Steel Dumont Tweezers (cat# RS-5045.Roboz, Gaithersburg, MD)
- Modified glass “transfer” pipettes (bottom of Pasteur pipette removed and edge smoothed)
- Aclar plastic film cut to 2″in x 2″ in squares (Honeywell, # 812071, Pottsville, PA)
- Cover sponges 4 in by 3 in ( Kendall , cat# 3157,Mansfield, MA)
- 30 mm Millicell membrane inserts (0.4 μm; Millipore, Bedford, MA)
- Mcllwain Tissue chopper with double-edged razor blade (McILwain tissue chopper, Vibratome Company, St. Louis, MO)
- Inverted microscope with CCD camera and image analysis software
(ii) Dissection Medium (DM; 1 liter at pH 7.3)
| Hank’s Balanced Salt | 1 vial ( 95.2 g/1L) ( Sigma : H2387 ) |
| NHCO3 ( 4.2 mM) | 350 mg |
| HEPES (10 mM) | 2.83 g |
| Glucose ( 33.3 mM) | 6.0 g |
| Penicillin/streptomycin (100x) | 10 mL |
| BSA (0.3%) | 300 mg |
| MgSO4-7H2O | 1.44 g |
(iii) Culture medium (400 mL)
| MEM (Invitrogen 11575-032) | 200 mL |
| ***Hank’s Balance Salt(Invitrogen 24020-117) | 100 mL |
| Horse serum (Invitrogen 26050-070) | 100 mL |
| HEPES buffer (1 M; Invitrogen 15630-080) | 5 mL |
| Penicillin/Streptomycin (100x) | 4 mL |
| ***Add 12.8 g glucose to 500 mL Hank’s Balance Salt before making culture medium | |
(iv) Animals
7 day old rat or mouse postnatal pups, generally one litter per experiment.
2. On the Day of Culture
- All dissection tools and Aclar film must be soaked in 70% ETOH for 30 minutes before beginning. The surface of the dissection hood, blade, and tissue chopper must be disinfected with ethanol.
- Prepare several (3-4) 6 well tissue culture plates; add 1 mL culture medium per well, and insert a Millipore permeable membrane in each well. Place 6-well plates in the 37° incubator until ready to transfer slices.
- Prepare several (4-6) 60 mm plates with DM and place on ice.
- Rapidly decapitate with scissors and dip in 70% ETOH, and quickly expose the brain with a saggital incision of the skull. Carefully remove the brain and place immediately in cold DM. Separate it into two hemispheres and place each hemisphere medial side down on a cover sponge for 2-3 seconds.
- Gently lift and place the hemisphere onto the Aclar film square and place on the tissue chopper; cut the brain coronally in 350 μm sections. When finished, gently take the Alcar square with the sectioned hemisphere and submerge in cold DM.
- Using a dissecting microscope, separate out the hippocampal sections. For rat, there should be 5 sections spanning rostral to caudal hippocampus, and for mouse there should be 3-4 sections.
- Take out the 6-well plates with membrane inserts from the 37° tissue culture incubator. Transfer hippocampal slices individually onto the permeable membranes using the glass transfer pipette. If too much DM is released onto the membrane, remove excess DM with a Pasteur pipette. Place five hippocampal slices on each membrane. Allow at least 3 wells per condition, or 15 slices.
- Incubate the culture plates at 37° in a 5% CO humidified incubator for 12-14 days before beginning the toxicity experiment. The culture medium can be changed twice a week.
3. Inducing and Quantifying Hippocampal Neuronal Injury in Slice Culture
- After 12-14 days in culture, add 5 μg/mL propidium iodide (PI) to the culture media. This step will allow visualization and quantification of basal cell death.
- The following day, quantify PI fluorescence in the CA1 subregion (and/or CA3 or dentate if desired) of each hippocampal slice and obtain mean the PI fluorescence measurements representing basal cell death (referred to as time=0 hours, or T0). For image acquisition and quantification, we use OpenLab software (Improvision, U.K.), but other image acquisition and analysis software can be used to quantify fluorescence intensity.
- Immediately after image acquisition of T0 PI fluorescence, induce neuronal injury by your method of choice (we have used 10 μM NMDA, oxygen-glucose deprivation, or LPS for one hour to induce neuronal toxicity 2,5). After the toxic stimulus, replace medium with fresh medium containing 5 μg/mL PI and maintain slices overnight.
- On day 3, quantify mean PI fluorescence of the experimental condition at 24h, or T24.
- After this image acquisition, immediately change medium and add 10 μM NMDA and 5 μg/mL PI and incubate slices overnight to achieve maximal neuronal death.
- On day 4, quantify mean PI fluorescence of maximal neuronal death, defined as Tmax. Calculate percent cell death as follows: (T24 – T0) / (Tmax-T0) .
4. Representative Results
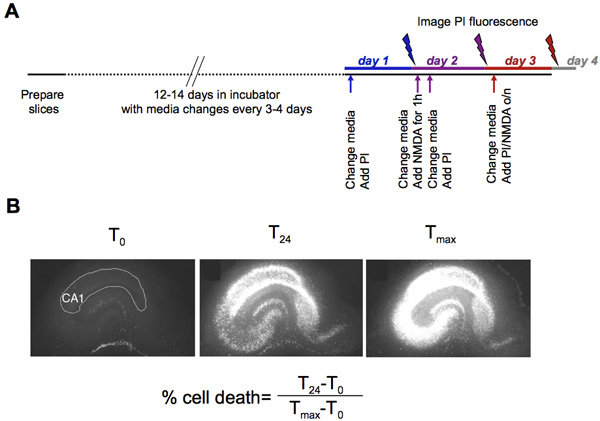
Figure 1. (A) Time line for generation of hippocampal and subsequent experimental injury and image acquisition. (B) Representative images of hippocampal slices basally (T0), 24 hours following a 1 hour exposure to 10 μM NMDA (T24), and after a further 24h or 10 μM NMDA to obtain maximal neuronal injury (Tmax).
