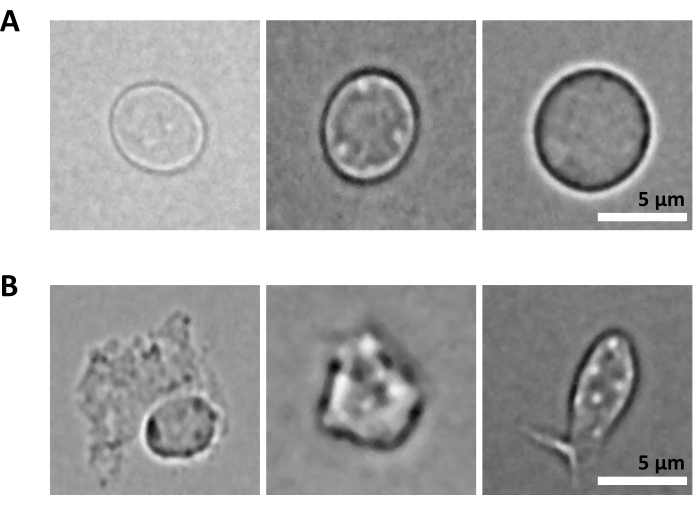
Les procédures expérimentales ont été menées dans le strict respect réglementaire des protocoles approuvés par le Comité d’éthique de l’expérimentation animale (CETEA). Pour l’isolement des noyaux cérébraux, des souris C57BL/6 âgées de 3 mois ont été utilisées. Pour l’isolement de la moelle osseuse, des souris femelles C57BL/6J âgées de 8 semaines pesant 18 g ont été utilisées. 1. Purification des noyaux du cerveau de souris REMARQUE : Portez des gants en latex ou en nitrile en tout temps pendant la procédure. Il est fortement conseillé d’avoir deux personnes pour réaliser l’expérience, pour que les étapes 1 à 3 (c’est-à-dire la préparation de la suspension de noyaux uniques) soient effectuées par une seule personne, et que l’étape 4 (c’est-à-dire la préparation du trieur) soit effectuée en parallèle par une autre personne. Étant donné que le protocole est très sensible au facteur temps, il est essentiel de minimiser le temps de traitement des échantillons en préparant le trieur dès que la suspension de noyaux uniques est préparée. Préparation des réactifs et des matériauxStérilisez soigneusement les outils de dissection en autoclave (à 121 °C pendant 20 min) et lavez-les à l’éthanol à 70% juste avant utilisation. Préparez une boîte de Pétri par échantillon, remplie de 2 à 3 mL de solution saline glacée 1x Dulbecco’s phosphate-buffered (DPBS). Refroidissez la microcentrifugeuse à 4 °C, remplissez un seau de glace et mettez l’homogénéisateur à dounce en verre sur de la glace. Préparer le tampon de lyse des noyaux en ajoutant de la digitonine pour obtenir une concentration finale de 0,01 %, 10 mL par échantillon. Préparer le tampon de coloration en ajoutant des inhibiteurs de la RNase au tampon de coloration cellulaire pour obtenir une concentration finale de 0,2 U/μL, 20 mL par échantillon. Préparer le DPBS 0,04 % BSA en ajoutant des inhibiteurs de la RNase pour une concentration finale de 0,2 U/μL, 2 mL par échantillon. Préparer 1 mL de tampon de noyaux dilués selon le protocole Multiome25. Conservez tous les réactifs et les échantillons sur de la glace. Dissection tissulaireSacrifiez les souris en utilisant des protocoles approuvés par l’établissement. Dans ce protocole, les souris ont été décapitées après une surdose de kétamine/xylazine. Coupez la tête de la souris avec des ciseaux et retirez le cerveau du crâne comme décrit dans Meyerhoff et al.26. Transférez immédiatement le cerveau dans une boîte de Pétri préparée avec le 1x DPBS glacé sous un stéréomicroscope éclairé par diodes électroluminescentes (LED). Coupez le tissu cérébral à l’aide d’un scalpel pour séparer les zones d’intérêt du cerveau (par exemple, le cortex entorhinal, l’hippocampe, le cortex préfrontal) et transférez chaque région dans une boîte de Pétri séparée contenant 1x DPBS glacé. Garder sur la glace. À l’aide d’un scalpel, émincez le tissu en morceaux de <0,5 cm pour faciliter l’homogénéisation à l’étape suivante. À l’aide d’une micropipette P1000, transférez le tissu haché et le 1x DPBS de la boîte de Pétri dans un tube de 1,5 ml. Assurez-vous d’utiliser des tubes en plastique à faible liaison aux protéines. Laissez les morceaux de tissu se séparer par gravité. Retirez délicatement l’excédent de 1x DPBS à l’aide d’une micropipette P1000.REMARQUE : Après cette étape, il est possible de congeler le tissu haché en transférant les tubes à faible liaison protéique sur de la glace sèche, puis en les stockant à -80 °C jusqu’à ce que l’on procède à l’isolement des noyaux. Isolement des noyauxRemplir la cuvette en verre avec 2 mL de tampon de lyse des noyaux glacés avec 0,01 % de digitonine. Ajoutez les morceaux de mouchoirs dans la dounce.REMARQUE : si vous travaillez avec des tissus frais congelés, ajoutez le tissu congelé haché directement dans le tampon de lyse des noyaux 0,01% digitonine ; Ne laissez pas le tissu décongeler avant. Homogénéiser à l’aide d’un homogénéisateur de tissu en verre 25 fois avec le pilon A, puis 25 fois avec le pilon B. Transférer l’homogénéat dans un tube de 15 mL. Ajouter 2 mL supplémentaires de tampon de lyse des noyaux glacés avec 0,01 % de digitonine et incuber sur glace pendant 5 minutes. Centrifuger les noyaux à 500 x g pendant 5 min à 4 °C. Retirer le surnageant à l’aide d’une micropipette et ajouter 4 mL de tampon de lyse des noyaux glacés contenant 0,01 % de digitonine. Incuber sur de la glace pendant 5 min et filtrer à travers une passoire à cellules de 40 μm. Centrifuger les noyaux à 500 x g pendant 5 min à 4 °C et retirer le surnageant à l’aide d’une micropipette. Ajouter 4 mL de tampon colorant pour laver les noyaux et centrifuger à 500 x g pendant 5 min à 4 °C. Retirer le surnageant à l’aide d’une micropipette et remettre la pastille en suspension dans 4 mL de tampon de coloration. Filtrer à travers une crépine de 40 μm et centrifuger à 500 x g pendant 5 min à 4 °C. Remettre en suspension dans 1 mL de PBS avec 0,04 % de BSA. Compter les noyaux pour assurer l’uniformité de la préparation des tissus/noyaux dans les différents échantillons. On s’attend à ce qu’il obtienne un nombre de noyaux similaire à partir des mêmes régions du cerveau :Ajouter 10 μL de bleu de trypan à 0,4 % dans un tube vide de 0,5 mL. Ajouter 10 μL de noyaux et mélanger 5 fois par pipetage. Compter les noyaux à l’aide d’un compteur de cellules automatisé en suivant les recommandations du fournisseur. Gardez les noyaux sur de la glace. Préparer les noyaux pour le tri.REMARQUE : Les noyaux extraits incorporent du 7-AAD, et cette coloration est utilisée pour leur purification par trieur de cellules activées par fluorescence (FACS).Transférez 100 μL de noyaux dans un tube FACS pour le contrôle non coloré. Ajouter 10 μL de 7-AAD aux noyaux restants et maintenir 5 min à 4 °C. Trier un minimum de 0,5 x 106 noyaux par FACS pour éliminer les doublets et les débris. Tri des noyaux à l’aide d’un FACSREMARQUE : Bien que le tri des noyaux puisse être effectué sur une grande variété de trieurs de cellules, la procédure d’utilisation des instruments BD FACSAria Fusion ou BD FACSAria III est décrite ici. Il est fortement recommandé que l’étalonnage et la configuration du trieur de cellules soient effectués sous la supervision ou par un utilisateur expérimenté de l’instrument. Pour réduire le temps de traitement des échantillons, il est essentiel que le trieur soit prêt dès que la suspension de noyaux uniques est préparée.Étalonnage de l’instrument FACSAllumez le trieur de cellules et l’ordinateur. Une fois le logiciel connecté à l’instrument, lancez la procédure de démarrage fluidique. Sélectionnez Cytomètre > démarrage fluidique dans le menu principal et suivez les quatre étapes. Cliquez sur Terminé après avoir terminé chacune d’entre elles. Insérez la buse de 70 μm, allumez le jet et laissez le jet se stabiliser pendant 15 min. Ajustez l’amplitude pour obtenir la formation de gouttes et cliquez sur Sweet Spot. Placez le filtre à densité neutre (N.D) 1.0 et ouvrez l’interface de configuration et de suivi du cytomètre (CST). Contrôle de qualité quotidien : Diluer les billes de CST dans un milieu FACS (voir les recommandations du fournisseur) et effectuer le contrôle CST. Une fois terminé, remplacez le N.D 1.0 par le N.D 2.0. Diluez Accudrops dans un milieu FACS (voir les recommandations du fournisseur) et effectuez un délai de goutte comme décrit aux étapes 6 à 10. Dans le modèle d’expérience, sélectionnez l’expérience Accudrop Drop Delay et ouvrez la mise en page de tri du tube. Dans la fenêtre inférieure de la caméra, cliquez sur Tension , puis sur Filtre optique pour activer l’application de la charge sur les plaques de déviation et l’utilisation d’un filtre optique spécifique devant la caméra. Assurez-vous que le quadrant sur le côté droit indique 100. Si nécessaire, ajustez la vis laser rouge pour optimiser l’impact du laser. Ajustez le débit pour atteindre la vitesse de 1 000 à 3 000 événements par seconde. Cliquez sur Trier et annuler. Assurez-vous que le quadrant gauche est égal à 100 et que le quadrant droit est égal à 0. Si le quadrant gauche est inférieur à 95, effectuez l’option Délai automatique. Cliquez sur Tension, puis sur Tri des tests. Contrôlez la qualité du dépôt des flux latéraux dans les tubes de collecte. Si nécessaire, ajustez la position des flux latéraux en déplaçant les curseurs. Mise en place de l’instrument FACS pour le tri des noyaux.Commencer l’acquisition des noyaux non colorés. Ceux-ci sont utilisés pour définir les diffusions directes et latérales, ainsi que la tension du détecteur pour le paramètre 7-ADD. Réglez les paramètres de manière à ce que le signal 7-AAD de l’échantillon non coloré se situe à l’intérieur de la première décade de l’échelle logarithmique sur le graphique à points. Commencez à acquérir le tube des noyaux colorés par 7-AAD et définissez les populations de noyaux en utilisant une stratégie de déclenchement basée sur (1) FSC-A/SCC-A puis FSC-H/SSC-H pour la taille et la granularité, (2) FSC-H/FSC-A pour la discrimination des doublets, et (3) SSC-A/7-AAD pour les noyaux positifs 7-AAD (voir Figure 2A). Assurez-vous que le jet et la déviation sont stables. Dans la caméra à flux latéral, activez le tri de test, la tension activée et confirmez le tri précis des gouttes dans un tube de 1,5 ml monté sur le côté gauche. Dans la fenêtre Mise en page de tri , sélectionnez la population d’intérêt, telle que définie à l’étape 2 (ci-dessus). Dans Evénements cibles, sélectionnez le seuil dans Continu pour obtenir un minimum de 0,5 x 106 noyaux par échantillon. Sous Précision, sélectionnez Pureté à 4 voies. Une fois prêt, cliquez sur Trier et sur OK pour commencer le tri des noyaux. Contrôle de la qualité et comptage des noyaux purifiésREMARQUE : Cette étape ne doit être effectuée que pendant l’expérience pilote pour l’optimisation des étapes de préparation des échantillons, dans le but de tester la pureté des noyaux triés qui doivent être chargés sur la puce de chrome 10X. Une fois que le protocole est entièrement optimisé, il n’est pas conseillé d’effectuer cette étape de contrôle de la qualité dans les expériences de suivi afin d’éviter le gaspillage inutile des noyaux collectés qui peuvent être disponibles en faible nombre.Contrôle de la pureté par cytométrie en fluxTransférer 10 μL des noyaux triés dans un nouveau tube FACS contenant 90 μL de DPBS avec 2 % de sérum de veau fœtal inactivé par la chaleur (HI-FBS). Acquérir et enregistrer les données post-tri pour vérifier la pureté et la viabilité du tri. S’assurer qu’au moins 98 % des noyaux apparaissent dans la porte d’intérêt, telle que définie à la section 4.2 (voir la figure 2B). Compter les noyaux purifiésCentrifuger les noyaux triés pendant 5 min à 500 x g et à 4 °C et retirer soigneusement le surnageant à l’aide d’une micropipette. Remise en suspension dans 100 μL de tampon de noyaux dilués. Ajouter 10 μL de bleu de trypan à 0,4 % dans un tube vide de 0,5 mL. Ajouter 10 μL des noyaux triés et mélanger 5x par pipetage. Compter les noyaux à l’aide d’un compteur de cellules automatisé en suivant les recommandations du fournisseur. Ajustez la concentration des noyaux à 3,5 x 106/mL, c’est-à-dire 16 000 noyaux par 5 μL. Contrôle de la qualité des noyaux purifiés par microscopieREMARQUE : Cette étape ne doit être effectuée que pendant l’expérience pilote pour optimiser les étapes de préparation des échantillons afin de tester la qualité des noyaux qui doivent être chargés sur la puce de chrome 10X. Une fois que le protocole est entièrement optimisé, il n’est pas conseillé d’effectuer cette étape de contrôle de la qualité dans les expériences de suivi afin d’éviter le gaspillage inutile des noyaux collectés qui peuvent être disponibles en faible nombre.Assurez-vous que les lames de microscope et les lamelles de recouvrement sont propres et exemptes de poussière. Si nécessaire, lavez et rincez les lamelles avec de l’éthanol absolu et séchez-les avec des lingettes non pelucheuses. Distribuer 25 μL de poly-l-lysine dans les puits coulissants qui seront utilisés et incuber pendant 10 min à température ambiante (RT), à l’abri de la poussière. Retirez l’excès de poly-l-lysine et ajoutez 10 μL de suspension de noyaux purifiés. Incuber pendant 5 min à RT, à l’abri de la poussière. Ajoutez une goutte de milieu de montage dans chaque puits, en évitant les bulles. Placez une lamelle au-dessus des puits ensemencés. Couvrir de lingettes en papier et appuyer fermement sur la lamelle pour retirer l’excédent de support de montage. Veillez à ne pas déplacer la lamelle et à ne pas nettoyer l’excès de support de montage. Prenez plusieurs images avec un microscope inversé avec une lumière à fond clair et un grossissement minimum de 40x. Effectuer un test multiome.Passez immédiatement au Guide de l’utilisateur de Chromium Next GEM Single Cell Multiome ATAC + Gene Expression (CG000338 – Rev F)25. 2. Purification des noyaux des cellules souches hématopoïétiques et progénitrices de la moelle osseuse de souris (HSPC) NOTE Ce protocole décrit la purification des noyaux de trois sous-ensembles de cellules souches hématopoïétiques (CSH) de la lignée c-Kit + Sca-1+ Sca-1+, des progéniteurs myéloïdes communs (CMP) de la lignée c-Kit + Sca-1-CD34 + FcγR – et de la lignée c-Kit + Sca-1-CD34 + FcγR+ progéniteurs granulocytes-monocytes (GMP). Portez des gants en latex ou en nitrile en tout temps pendant la procédure. Ce protocole est une adaptation du 10X Genomics Demonstration Protocol – Nuclei Isolation for Single Cell Multiome ATAC + GEX sequencing (CG000365 – Rev C)27. Des modifications ont été introduites dans le protocole original afin de maximiser la récupération des noyaux. Il est fortement conseillé d’avoir deux personnes qui réalisent l’expérience, pour avoir les étapes 1. à 3. (c’est-à-dire la préparation de la solution unicellulaire) effectuée par une personne, et l’étape 4 (c’est-à-dire la préparation du trieur) effectuée en parallèle par une autre personne. Étant donné que le protocole est très sensible au facteur temps, il est essentiel de minimiser le temps de traitement des échantillons en préparant le trieur dès que la suspension à cellule unique est préparée. Préparation des réactifs et des matériauxRemplissez un seau de glaçons. Préparer le tampon FACS : DPBS avec une solution HI-FBS à 2 % (environ 500 mL pour 6 échantillons) et filtrer à travers un filtre de 0,2 μm. Préparer le milieu de prélèvement : DPBS avec une solution HI-FBS à 10 % (500 μL par échantillon) et filtrer à travers un filtre de 0,2 μm. Isolement des cellules de la moelle osseuseSacrifiez les souris en utilisant des protocoles approuvés par l’établissement. Dans cette expérience, les souris ont été sacrifiées par luxation cervicale après une surdose de kétamine/xylazine. Vaporisez l’abdomen et les pattes arrière des souris avec 70 % d’éthanol. À l’aide d’une pince et de ciseaux stériles, faites une petite incision au milieu du bas-ventre et ouvrez le péritoine de la base des pattes postérieures jusqu’au diaphragme (figure supplémentaire 1). Faites une incision supplémentaire pour chaque patte arrière perpendiculaire au péritoine ouvert, puis saisissez l’un ou l’autre côté de l’une de ces entailles supplémentaires et écartez-la pour décoller la peau des deux pattes arrière au-delà de l’articulation de la cheville afin d’exposer les muscles des deux pattes arrière (figure supplémentaire 1A). Alignez les ciseaux le long de la colonne vertébrale au niveau de l’articulation de la hanche d’une patte arrière pour couper la patte sans couper le fémur (figure supplémentaire 1B, C). Répétez la même chose pour l’autre jambe. Pour isoler le fémur, découpez la majeure partie du tissu musculaire, puis tenez le fémur et le tibia dans chaque main avec le bout des doigts au niveau de l’articulation (figure supplémentaire 1D, E). Pliez doucement la jambe contre la courbure naturelle pour disloquer le tibia du fémur (figure supplémentaire 1E), puis coupez soigneusement le tissu conjonctif avec des ciseaux pour séparer le fémur et le tibia. Utilisez les ciseaux avec de légers mouvements de torsion pour disloquer le morceau de colonne vertébrale de l’extrémité supérieure du fémur (figure supplémentaire 1E). Nettoyez le fémur isolé avec du papier de soie pour enlever le muscle et le tissu conjonctif restants. Conserver au froid sur de la glace dans un puits à plaque de 12 puits rempli de 2 mL de DMEM (1x) + GlutaMAX-I. Une fois que tous les fémurs sont prélevés, assurez-vous que les tissus musculaires et fibreux sont complètement retirés de l’os. Ne coupez pas l’os pour (a) garder la moelle à l’intérieur stérile et (b) éviter de perdre des cellules dans le puits. Utilisez les étapes suivantes pour rincer les cellules de deux fémurs d’une souris, adaptées de Haag et Murthy28. Préparez un tube de 1,5 ml et un tube de 0,5 ml. Ajoutez 150 μL du tampon FACS dans le tube de 1,5 mL, puis percez un trou au fond du tube de 0,5 mL à l’aide d’une aiguille de 18 G et insérez le tube de 0,5 mL dans le tube de 1,5 mL. Ouvrez la partie distale de chaque fémur à l’aide de ciseaux chirurgicaux de souris (Figure supplémentaire 1F) : Verrouillez l’épiphyse distale entre les lames et appliquez une légère pression tout en retournant les ciseaux pour détacher en douceur l’épiphyse distale sans ouvrir l’os durement. En cas de succès, 4 protubérances devraient être visibles à l’extrémité de la physe maintenant exposée (figure supplémentaire 1G). Insérez les deux fémurs avec l’extrémité ouverte vers le bas dans le tube préparé de 0,5 mL placé à l’intérieur d’un tube de 1,5 mL contenant un tampon FACS (Figure supplémentaire 1H). Placez une crépine de 70 μm sur un tube de 50 ml et prémouillez la crépine avec 2 mL de tampon FACS. Pour rincer la moelle osseuse, centrifugez les tubes (bouchons ouverts) à 12 000 x g jusqu’à ce que la centrifugeuse atteigne la valeur de 12 000 x g , puis arrêtez immédiatement la centrifugeuse. Vérifier que les cellules de la moelle osseuse sont enrobées dans le tube de 1,5 mL et que les fémurs sont blancs (avant le rinçage des cellules, ils sont rouges) (Figure supplémentaire 1I). Jeter les tubes de 0,5 ml avec les 2 fémurs. Jeter le surnageant de 150 μL à l’aide d’une pipette. Remettre la pastille en suspension à l’aide d’une micropipette dans 1 mL de tampon de lyse au chlorure d’ammonium-potassium (ACK) pendant 1 à 2 minutes à la température RT pour lyser les globules rouges. Évitez les temps d’incubation plus longs car ils peuvent entraîner une diminution de la viabilité des cellules nucléées. Transvaser dans le tube de 50 mL à travers la crépine de cellules pré-humidifiée de 70 μm. Ajouter 10 mL de tampon FACS pour diluer le tampon de lyse ACK et ainsi arrêter la lyse. Centrifuger à 400 x g pendant 5 min à 4 °C. Remettre en suspension dans 10 mL de tampon FACS en remettant d’abord en suspension dans 1 mL, puis en complétant avec 9 mL. Préparez les cellules pour le comptage comme décrit au point 1.3.8. Comptez les cellules à l’aide d’un compteur de cellules automatisé en suivant les recommandations du fournisseur. On s’attend à ce qu’il prélève environ 40 millions de cellules de 2 fémurs. Coloration de la moelle osseuse HSPCCentrifuger les cellules à 400 x g pendant 5 min à 4 °C et remettre en suspension la pastille à l’aide d’une micropipette dans un tampon FACS jusqu’à une concentration finale de 1 x 107 cellules/mL. À l’aide d’une micropipette P1000, transférez la suspension dans un tube FACS, en filtrant à travers un capuchon de filtre à cellules de 35 μm. Préparez des échantillons de tube à essai à coloration unique pour chaque anticorps répertorié dans le tableau 1 afin de mettre en place des compensations de fluorochromes sur le trieur de cellules :Préparez un tube FACS par anticorps et remplissez les tubes avec 200 μL de PBS. Ajouter 15 μL de billes de compensation fluorochrome dans chaque tube FACS d’anticorps conjugué au fluorochrome. Dans les tubes FACS pour les cellules colorées simples non colorées et pour les cellules colorées vivantes/mortes, ajoutez 500 000 cellules au lieu de billes. Ajouter 1 μL de chaque anticorps conjugué au fluorochrome (voir le tableau 1) dans le tube FACS correspondant. Ajouter 0,5 μL de colorant vivant/mort dans le tube FACS à coloration unique vivant/mort. Conserver sur glace pendant 15 min à l’abri de la lumière. Préparer les mélanges 1 et 2 comme indiqué dans le tableau 2.REMARQUE : Les volumes d’anticorps indiqués dans le tableau 2 sont valides pour les anticorps référencés dans le tableau des matériaux. Ils doivent être optimisés pour toute nouvelle référence d’anticorps ou un lot différent de la même référence d’anticorps. Ajouter 300 μL de Mix 1 dans le tube d’échantillon, remettre en suspension et conserver pendant 15 minutes sur de la glace à l’abri de la lumière. Ajouter 300 μL de Mix 2 dans le tube d’échantillon, remettre en suspension et conserver pendant 20 minutes sur de la glace à l’abri de la lumière. Ajouter 3 mL de tampon FACS dans les tubes à coloration unique et les tubes d’échantillon colorés au mélange. Essorer à 400 x g pendant 5 min à 4°C. Jeter délicatement le surnageant à l’aide d’une micropipette et remettre la pastille en suspension dans 500 μL du tampon FACS. Préparez un tube de 1,5 mL prérempli de 500 μL de milieu de collecte.REMARQUE : Le mélange 1 est préparé dans le DPBS car il contient la coloration vivante/morte significativement affectée par HI-FBS. Une fois que les cellules sont colorées par Live/Dead, le mélange 2 est ajouté, qui contient les anticorps conjugués au fluorochrome remis en suspension dans un tampon FACS contenant HI-FBS. La seule exception est l’anticorps anti-CD16/32 qui est inclus dans le mélange d’anticorps 1 pour servir de bloqueur des récepteurs Fc qui empêche la liaison non spécifique des autres anticorps ajoutés à l’étape suivante. Tri des cellules à l’aide d’un FACSREMARQUE : Bien que le tri cellulaire puisse être effectué sur une grande variété de trieurs de cellules, la procédure d’utilisation des instruments BD FACSAria Fusion ou BD FACSAria III est décrite ici. Il est fortement recommandé que l’étalonnage et la configuration du trieur de cellules soient effectués sous supervision ou par un utilisateur expérimenté de l’instrument.Étalonnage de l’instrument FACS : Reportez-vous à l’étape 4.1 du protocole 1. Mise en place de l’instrument FACS pour le tri des cellules :Commencez l’acquisition des cellules non colorées. Ceux-ci sont utilisés pour définir les diffusions directes et latérales et la tension du détecteur pour chaque fluorophore. Définissez les paramètres de manière à ce que le signal fluorescent de chaque fluorophore se situe à l’intérieur de la première décade de l’échelle logarithmique sur le graphique à points. Acquérez des contrôles de couleur unique pour configurer manuellement les compensations (la médiane des populations positives et négatives doit être alignée) ou utilisez le logiciel de calcul automatique (mesures de pente). Assurez-vous que les commandes de compensation correspondent aux fluorochromes expérimentaux et aux paramètres du détecteur. Enregistrez 10 000 événements pour les cellules et 5 000 événements pour les perles. Utilisez le tube d’échantillon (c’est-à-dire les cellules multi-colorées) pour définir les populations cellulaires d’intérêt en utilisant la stratégie de déclenchement illustrée à la figure 3A. Suivez les étapes 4 à 6 (ci-dessous). Pour identifier les trois HSPC de moelle osseuse d’intérêt (HSC, CMP et GMP), commencez le déclenchement en utilisant la taille (FSC-A) et la granularité (SSC-A) pour porter sur les leucocytes, puis FSC-H/FSC-A pour discriminer les doublets. Sur la base du marqueur SSC-A/cellules mortes, porte les cellules vivantes. Utilisez Lineage/c-Kit pour sélectionner des cellules qui sont négatives pour la lignée et qui expriment des niveaux intermédiaires à élevés de c-Kit. Par l’intermédiaire de c-Kit/Sca-1, porte sur les CSH de la lignée-c-Kit+ Sca-1+ (LKS+), l’une des trois populations d’intérêt. Parmi les progéniteurs myéloïdes (lignée-c-Kit+Sca-1-), utiliser FcγR/CD34 pour exclure les progéniteurs mégacaryocytaires et érythroïdes (MEP) CD34-FcγR- tout en incluant CD34+ FcγR- CMP, ainsi que CD34+FcγR+ GMP dans les populations cellulaires à trier. Assurez-vous que le jet et la déviation sont stables. Dans la caméra à flux latéral, activez le tri de test, la tension activée et confirmez le tri précis des gouttes dans un tube de 1,5 ml monté sur le côté gauche. Dans la fenêtre Disposition de tri , sélectionnez la ou les populations d’intérêt (c’est-à-dire « LKS+ » et « progéniteurs myéloïdes CD34+ » illustrés dans cet exemple). Sous Appareil, sélectionnez 2 tubes. Sous Précision, sélectionnez Pureté. Dans Événements cibles, sélectionnez Continu pour trier entre 160 000 et 200 000 progéniteurs myéloïdes LKS+ et CD34+ . Ajouter 500 μL de tampon FACS à la suspension cellulaire et transférer 1 mL de l’échantillon en le filtrant dans un nouveau tube FACS à couvercle de crépine de 35 μm pour s’assurer que toutes les cellules sont dans une seule suspension juste avant l’acquisition. Cela permet d’éliminer les amas de cellules qui pourraient obstruer l’instrument. Une fois prêt, cliquez sur Trier et sur OK pour lancer le tri. Ajustez le débit pour maintenir la vitesse en dessous de 10 000 événements par seconde.REMARQUE : Le rapport attendu entre les progéniteurs myéloïdes LKS+ et CD34+ est de 1 :3 pour une souris femelle C57BL/6J adulte (âgée de 8 à 12 semaines) à l’état d’équilibre. Les numéros de cellules triées ciblés sont généralement atteints dans les 30 minutes suivant le tri. Contrôle de la qualité et comptage des cellules triéesREMARQUE : Cette étape ne doit être effectuée que pendant l’expérience pilote d’optimisation des étapes de préparation des échantillons, dans le but de tester la pureté des cellules triées qui doivent être utilisées pour l’isolement des noyaux. Une fois le protocole entièrement optimisé, il n’est pas conseillé d’effectuer cette étape de contrôle de la qualité dans les expériences de suivi afin d’éviter le gaspillage inutile de matière première qui peut être disponible en faible nombre pour l’isolement des noyaux.Contrôle de la pureté par cytométrie en fluxTransférer 10 μL des cellules triées dans un nouveau tube FACS contenant 90 μL de tampon FACS. Acquérir et enregistrer les données post-tri pour vérifier la pureté et la viabilité du tri. Assurez-vous qu’au moins 95 % des cellules apparaissent dans la porte d’intérêt, telle que définie aux points 3 à 6 et illustrée à la figure 3B. Isolement de noyaux à partir de HSPC triés dans la moelle osseuseUtiliser le protocole « Low Cell Input Nuclei Isolation » de l’annexe du 10X Genomics Demonstration Protocol – Nuclei Isolation for Single Cell Multiome ATAC + GEX sequencing (CG000365 – Rev C)27, avec les modifications suivantes apportées pour optimiser la récupération des noyaux :Temps de lyse : Effectuez une expérience pilote pour ce protocole afin d’identifier le meilleur moment de lyse pour l’isolement des noyaux. Assurez-vous d’obtenir une lyse cellulaire complète tout en maintenant des noyaux intacts.REMARQUE : L’étape f du protocole 10X Genomics27 mentionné ci-dessus indique de « incuber [dans un tampon de lyse] pendant 3 à 5 minutes sur de la glace ». Au cours de l’expérience pilote, testez au moins pendant 3 min, 4 min et 5 min et évaluez la quantité de noyaux récupérés par comptage et qualité par cytométrie en flux et imagerie microscopique pour choisir la durée optimale de lyse (voir la description de ces contrôles de qualité ci-dessous). Pour épargner les réactifs, remplacer le tampon de noyaux dilués par du PBS 0,04 % BSA dans l’expérience pilote. Pour les HSPC de la moelle osseuse, 3 min ont été identifiés comme la durée optimale de la lyse. Centrifugations cellulaires : Pour toutes les centrifugations cellulaires en suspension, centrifuger à 300 x g pendant 7 min (au lieu des 5 min en CG000365 – Rev C)27 à 4 °C. Centrifugations des noyaux : Effectuer toutes les centrifugations en suspension des noyaux à 500 x g pendant 5 min conformément à CG000365 – Rev C27. Prélèvement des noyaux : À l’étape b, après avoir remis en suspension 50 μL de PBS 0,04 % BSA et transféré dans un tube de 0,2 mL, ajouter 50 μL de PBS 0,04 % BSA dans le tube d’origine et mélanger la pipette pour recueillir les cellules restantes. Transférer dans le tube de 0,2 mL pour atteindre un volume total de 100 μL. Dorénavant, le volume total sera de 100 μL au lieu des 50 μL du protocole. Ajustez les étapes en aval en conséquence (p. ex., pour l’étape d, retirer 90 μL au lieu de 45 μL ; pour l’étape e, ajouter 90 μL de tampon de lyse au lieu de 45 μL). Pour l’étape m, remettre en suspension la pastille de noyaux dans 12 μL de tampon de noyaux dilués au lieu de 7 μL. Comptez les noyaux isolés. Dans un tube vide de 0,5 mL, ajouter 10 μL de bleu de trypan à 0,4 % et 8 μL de PBS à 0,04 % BSA. Ajouter 2 μL de noyaux dans le tube et compter les noyaux comme décrit au point 1.3.8. Utilisez un compteur de cellules automatisé en suivant les recommandations du fournisseur. Contrôle de la pureté par cytométrie en fluxREMARQUE : Cette étape ne doit être effectuée que pendant l’expérience pilote pour optimiser les étapes de préparation des échantillons afin de tester la pureté des noyaux qui doivent être chargés sur la puce de chrome 10X. Une fois que le protocole est entièrement optimisé, il n’est pas conseillé d’effectuer cette étape de contrôle de la qualité dans les expériences de suivi afin d’éviter le gaspillage inutile des noyaux collectés qui peuvent être disponibles en faible nombre.Après avoir terminé l’isolement des noyaux, transférer 6 μL de noyaux remis en suspension dans un nouveau tube FACS prérempli de 150 μL de tampon FACS. Ajouter 3 μL de 7-AAD et incuber 5 min sur glace. Acquérir et enregistrer les données post-tri pour vérifier la pureté et la viabilité du tri. S’assurer qu’au moins 95 % des noyaux apparaissent dans la porte d’intérêt, tel que défini à l’étape 4.2 du protocole 1 (voir la figure 4). Contrôle de la qualité des noyaux purifiés par microscopie :REMARQUE : Cette étape ne doit être effectuée que pendant l’expérience pilote pour l’optimisation des étapes de préparation des échantillons afin de tester la qualité des noyaux qui doivent être chargés sur la puce de chrome 10X. Une fois que le protocole est entièrement optimisé, il n’est pas conseillé d’effectuer cette étape de contrôle de la qualité dans les expériences de suivi afin d’éviter le gaspillage inutile des noyaux collectés qui peuvent être disponibles en faible nombre.Procédez comme décrit à l’étape 1.5.3. Effectuer un test multiomePassez immédiatement au Guide de l’utilisateur de Chromium Next GEM Single Cell Multiome ATAC + Gene Expression (CG000338 – Rev F)25.