The design effort was a result of significant planning. Novel to this project is the use of alternative splicing to create a pre-mRNA that is translated into differentially expressed proteins. These proteins are expressed in different organelles, in this case the chloroplast, peroxisome and/or cytosol. We adapted natural Arabidopsis gene that is alternatively spliced6, and placed known chloroplast6 and peroxisome17 targeting sequences in alternate exons (TriTag-1 and TriTag-2). TriTag-3 consists of a peroxisome signal embedded within a low-complexity region of the chloroplast sequence. These were placed in frame with GFP (Figure 1).
The plasmids used in this study thus contained several elements: the native alternative splicing regimen from the PIMT2 gene6, chloroplast and peroxisome targeting sequences, GFP, and a plasmid backbone. Since each has very specific required sequence, a sequence-independent method is necessary. The Gibson assembly protocol12,13 can be used on any sequences so long as the constituent parts are designed with sequence similarity to each other, and do not contain repeating sequences. In brief, linear DNA fragments are constructed (either via PCR amplification, commercial synthesis, or digestion of a pre-made plasmid) such that they have ~50 bp of overlapping homologous sequence (Figure 2). Equimolar quantities of each of the DNA fragments are combined in a single tube containing the Gibson assembly mix, incubated at 50 °C for one hour, and transformed into E. coli. The plasmids resulting from the Gibson assembly protocol were confirmed by Sanger sequencing.
In this study the three different GFP organelle-targeting constructs were compared to untagged GFP and a Rubisco-tagged GFP. Confocal microscopy was used to detect GFP fluorescence in the single cells transiently transformed. In control experiments, the transient expression observed was correlated to the chloroplasts (easily identifiable by the red autofluorescence of chlorophyll), the nuclei (large organelle that is not a chloroplast and has only one per cell), and/or small organelles believed to be peroxisomes. Green (GFP) and red (chlorophyll autofluorescence) channels were overlaid to study intracellular localization (Figure 3). For a more complete discussion of these results, please see our companion article11.
As predicted, in all three dual targeting constructs, GFP was observed in the chloroplasts (Figure 4) in overlay confocal micrographs. TriTag-1 and TriTag-2 target the GFP also to the cytosol and the small organelle believed to be the peroxisome (Figures 4a and 4b, respectively). TriTag-3 targets the GFP to only the chloroplast and the peroxisome, but not the cytosol (Figure 4c). A summary of the results is diagrammed (Figure 5).
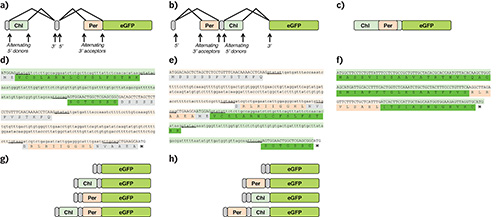
Figure 1. Design of alternative splicing constructs for differential organelle expression in Nicotiana benthamiana. Splice diagrams of (a) TriTag-1, (b) TriTag-2, and (c) TriTag-3 showing non-targeting sequences (gray), chloroplast targeting sequences (light green), peroxisome targeting sequences (orange), and the GFP coding sequence used in transient expression experiments. TriTag-1 and TriTag-2 consist of alternative splicing constructs; TriTag-3 consists of a peroxisome signal embedded within a low-complexity region of the chloroplast sequence. Sequences of (d) TriTag-1, (e) TriTag-2, and (f) TriTag-3. The ATG codon at the end of these sequences corresponds to the GFP start codon. Alternatively spliced targeting regions are underlined. Donor and acceptor dimers are underlined. The light green DNA sequences derive from the PIMT2 5’ coding region6 and include sequences encoding the chloroplast targeting sequence (green). The light orange DNA sequences derive from the TTL 5’ coding region17 and include sequences encoding the peroxisome targeting sequence (orange). TriTag-3 consists of a peroxisome signal embedded within a low-complexity region of the chloroplast sequence. Schematics of final mRNAs believed to be expressed for (g) TriTag-1 and (h) TriTag-2. Reprinted with permission11. Click here to view larger image.
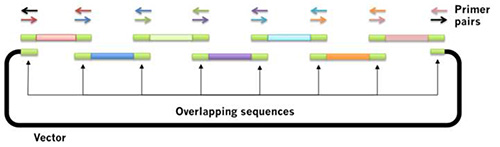
Figure 2. Schematic for Gibson assembly: a novel method for assembly of overlapping DNA fragments. The Gibson assembly method for plasmid construction12,13 is rapidly replacing traditional restriction-ligation cloning in many laboratories. Essentially, one can design a construct in silico that contains the DNA of interest exactly, composed of PCR products amplified from various sources. Design primers that are complimentary to each other, and PCR-amplify each fragment with the colored primers. Add all the pieces into a single tube with the enzyme mix, and transform E. coli competent cells. Click here to view larger image.
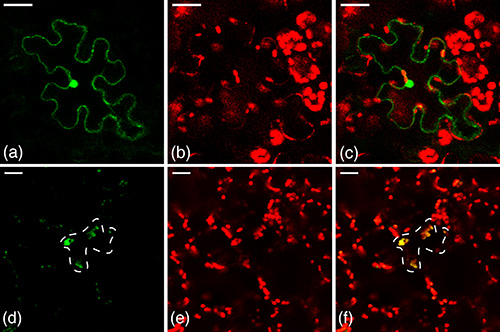
Figure 3. Control treatments bombarded into N. benthamiana leaf epidermal cells. (a-c) Untagged GFP. (a) Green channel. Untagged GFP is expressed in the nucleus and around the cell periphery, corresponding to the cytosol, but not the vacuole. (b) Red chloroplasts indicate autofluorescence of chlorophyll. (c) Overlay. In this construct fluorescence is observed in the nucleus and cytosol only, but not the chloroplasts. (d-f) Chloroplast-tagged GFP control. (d) Green channel. GFP is expressed in the large organelles, with a cell as outlined. (e) Red chloroplasts indicate autofluorescence of chlorophyll. (f) Overlay. In this case yellow chloroplasts are observed, indicating that the GFP (green) is targeted to the chloroplast. Reprinted with permission11. Click here to view larger image.
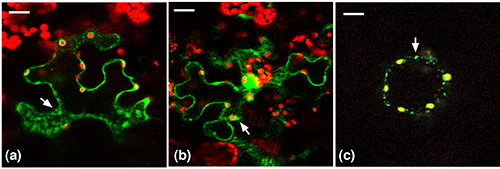
Figure 4. Tri-Tag-1, 2, 3 tagged GFP merged images. (a) Tri-Tag1. In this case yellow chloroplast peripheries are observed, indicating that the GFP is targeted to the chloroplast. GFP is also observed in the cytosol as well as small punctate organelles that may be peroxisomes. (b) Tri-Tag2. In this case yellow chloroplast peripheries are observed, indicating that the GFP is targeted to the chloroplast. GFP is also observed in the cytosol as well as small punctate organelles that may be peroxisomes. (c) Tri-Tag3. In this case yellow chloroplasts are observed, as well as small punctate organelles that may be peroxisomes. But GFP is not observed in the cytosol. Reprinted with permission11. Click here to view larger image.
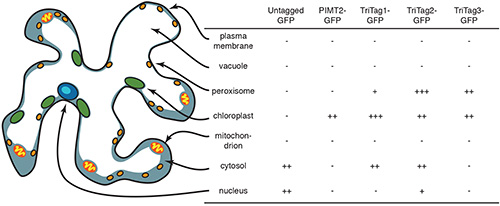
Figure 5. Compartments of a typical tobacco leaf epidermal cell. Here their relative sizes and locations within the cell are shown, and the relative expression levels observed via confocal microscopy. Reprinted with permission11. Click here to view larger image.
