


Summary
この手順では、スパイラル輪部とらせん神経節ニューロンの有無にかかわらず、コルチ器のマウス器官の単離および培養のための方法を説明します。我々はまた、エレクトロポレーションにより、コルチ植の器官における外因性レポーター遺伝子の発現のための方法を示しています。
Abstract
すべての哺乳類では、オーディションのための感覚上皮は、内耳の巻き貝の形をした蝸牛(図1)内に存在するコルチ器のスパイラル器官に沿って位置しています。聴覚系のmechanosensory細胞である発展途上蝸牛、、の有毛細胞は、1つの内側の有毛細胞の行で、3(ベースと半ばターンで)〜4(頂回転で)外有毛細胞の行に整列されていますそのスパンコルチ器官の長さを。有毛細胞は、脳が解釈できる神経インパルスに基底膜の音誘発性機械的振動を伝達する。感音難聴の症例の大部分は蝸牛有毛細胞の死や機能異常によって引き起こされます。
聴覚の研究にますます不可欠なツールでは、臓器の外植片1,2,9のin vitro培養における分離となります。一旦単離さ、外植片は、規範的、異常、または治療生理に関する情報を提供するために、いくつかの方法で利用することができる。遺伝子発現、不動の運動性、細胞および分子生物学だけでなく、有毛細胞の再生のための生物学的なアプローチは、コルチ器の外植片の器官の実験的なアプリケーションの例です。
このプロトコルは、新生児マウスからのコルチ器官の単離と培養のための方法を説明します。付属のビデオでは、仔マウス、および蝸牛のその後の分離、らせん靱帯、およびコルチ器官から側頭骨の分離のための段階的な指示が含まれています。一旦単離さ、感覚上皮が播種し、培養し、全体としてin vitroで 、または、さらに解剖スパイラル角膜輪部とらせん神経節の神経細胞を欠いているマイクロは、隔離とすることができます。この方法を使用して、一次外植片は7〜10日のために維持することができます。この手順のユーティリティの例として、コルチの外植片の器官は、外因性DsRedをレポーター遺伝子を電気穿孔される。それは分離、顕微解剖、およびコルチ器官の初代培養のための、再現性の明白な、と段階的な方向性を提供するため、このメソッドは、他の公開方法に比べて改善されています。
Protocol
1日目。ガラス製カバースリップの殺菌、コーティング。
- オートクレーブにガラスの顕微鏡のカバーグラスを滅菌乾燥させます。
- あらかじめ滅菌四ウェル細胞培養皿の二つウェルに滅菌カバーグラスを置きます。
- 午前1時01 ポリ- L -オルニチンとラミニンでカバースリップは、4℃で一晩20% ウシ胎児血清(FBS)を添加したコート℃に
- 400μLポリ- L -オルニチン(4で保存されている0.01%のソリューション° C)
- 400μLラミニン(-20アリコートに保存されて50μg/ mlのストック溶液° C)
- 200μLFBS(-20℃でアリコートに保存されている)
- 150℃インキュベーターで解剖ツールが一晩熱滅菌する。
- 10%の血清および10 mg / mLのアンピシリンを含む培地を加えます。
- 90mLのダルベッコ改変イーグル培地
- 5 mLのFBS(-20℃でアリコートに保存されている)
- 5mLのウマ血清 (-20℃でアリコートに保存されている)
- 10μL アンピシリン (4℃で保存されて10 mg / mlのストック溶液° C)
2日目。コルチ器官の単離。
- 正流の解剖のフードを滅菌する 。
- 20分間UVライトをオンにする
- 70%エタノールですべての面をスプレーし、使用前に5分を待つ。
- オペレーティングはさみを使用して大後頭孔の基部に子犬マウスを(P4)首を切る。
- 簡単に言うと70%エタノールを含む10cmディッシュで頭を洗う。
- 手術用メスの刃を使用して表皮を削除します。
- 手術用メスの刃を使用して矢状縫合に沿って頭蓋を開いて、前脳を二等分する。さらに解剖のための尾側前脳を維持する。
- 鈍的切開を用いて前脳、小脳と脳幹を削除します。
- 側頭骨(図2A)を削除し、70%エタノールで簡単にそれらを浸し、そして滅菌HBSSを含む3 mmディッシュに移す。
- 鉗子を使用して 、側頭骨錐体部から水疱と周囲の組織を削除します。
- 巻き貝形の蝸牛(図2B)を見つけて、ピンセットを使用して前庭系から分離する。
- 開発のこの段階では、骨迷路は完全に石灰化されていないと簡単に鉗子を用いて解剖される。基端から開始し、鉗子を使用してapically移動慎重に分離による蝸牛の骨迷路を削除します。
- らせん靱帯とコルチ器の接続された器官は蝸牛軸(図2C)のスパイラルに沿って巻かれている。慎重にピンセットを使用して、ベースのフックの領域でらせん靱帯を確保し、apically移動すると、それをほどくことによってコルチ器官を取り除く。
- ベースから始まる、 第55細かい鉗子 (図2D)を使用してコルチ器官かららせん靱帯を削除します。
コルティ感覚上皮(オプション)の臓器のマイクロ分離。 - 2つを使用して、ベースのコルティフック領域の臓器を取り除く½ ccのインスリン用注射筒を恒久的に接続されているU - 100 28G ½鉗子のような針で。
- 頂点から開始し、内有毛細胞の行からスパイラル輪部を削除し、(図2E - F)basally進みます。
コルチ器外植片の器官をめっき - 培養ウェルからpoly-L-ornithine/laminin/FBS溶液を除去し、培養液の130μlを加える。
- よくと東洋文化の中でコーティングされたガラスのカバースリップ有毛細胞の繊毛が上を向くように外植片にコルチ器の解剖臓器を転送する。
- 200μLピペットを用いて培養ウェルの培養液を除去。基底膜がコーティングされたガラスのカバースリップで固体接触することを確認してください。
- 慎重に200 mLのピペットを用いてコルチ器官に培養液の130μLの。コルチ器官の表面に2滴を適用してから、ゆっくりとカバースリップの側に、残りのボリュームを追加します。コルチ器官が培地に浮遊するが、カバースリップに貼られたままにしていないことを確認してください。
- 37 5%CO 2存在下で° Cで一晩インキュベートする。
3日目。コルチ器の培養臓器へのレポーター遺伝子のエレクトロポレーション。
- コルチ器の文化の臓器から培養液を取り外します。
- 1分を130μLH 2 Oを追加してから、200μLピペットを使用して削除します。
- プラスミドDsRedをレポーター(-20℃で保存2mgを/ mLのH 2 O)の30μLを追加。
- ようにマイクロマニピュレーターを用いてエレクトロの電極を進めるアノードNDカソードは、文化のいずれかの側にあります。
- コルティの移植片培養の臓器にレポーター遺伝子をエレクトロにパルス(27V、30ミリ秒持続時間、10種類のパルストレイン)を生成する。
- オプション:modiolarと植のらせん靱帯の両側に、導入遺伝子のエレクトロポレーションを確保するためにパルスの極性を逆に。
- 5分待ってください。
- DNA溶液 (2部DNAのトランスフェ3部):のFugene 6 130μLを加える。このソリューションは、前の層流フードで3 mLの丸底ポリスチレン試験管を用いてエレクトロポレーションの手順(ステップ20)の開始に準備する必要があります。このソリューションを作成するには:
- 試験管にのOpti - MEM(4℃で保存)2.4μLを加える。
- 試験管にのFugene 6試薬0.6μLを(4℃で保存)を追加します。のOpti - MEMに直接トランスフェを追加し、試験管の側面との直接接触を避けるようにしてください。
- 1秒間ボルテックスする。
- 室温で5分間インキュベートする。
- 2.0μLDsRedをレポータープラスミド二本鎖DNA(100μgのDNA / mLのH 2 Oのアリコートで-20℃で保存)を追加します。
- 1秒間ボルテックスする。
- 室温で15分間インキュベートする。
- 培養液200μLを加える。
- 1秒間ボルテックスする。
- ° C、5%CO 2で37℃で一晩インキュベートする。
- ウェル培養に2mLの培養液を追加し、37℃で10日までインキュベートする。
代表的な結果
我々は、周産期のマウスからのコルチ器官の単離のための方法を提示する。手順は、骨迷路が十分に解離が面倒なレンダリングに石灰化になる時点で、生後約最大16日胚のように若いマウス6で使用することができます。コルチ器官が解剖されると、それは全体(図3)やマイクロ分離された感覚上皮(図4)のいずれかで播種し、培養することができる。今後は、コルチ器の培養器官で外来遺伝子を表現する手法を提示している。器官培養は、らせん神経節細胞またはマイクロ分離を用いた外因性幹細胞とコルチ器官共培養、そのようなRT - PCRを用いてコルチ器の遺伝子発現の臓器の分析として、またはin situハイブリダイゼーション研究の他の多くの種類、のに便利です。 3、または有毛細胞の死と再生のインビトロ分析インチ

図1。コルチ器のP4マウス臓器の断面。 (A)P4マウスから得られたcryosectioned蝸牛の基底回転からのクロスセクションでは、このプロトコルで説明されているマウス蝸牛の一般的な構造を示しています。中央階は、下方スパイラル靭帯と横方向に血管条、ライスナーの上方に膜、内側螺旋縁、および基底膜に接しています。ボックスには、Bに拡大領域が(B)コルチ器官が基底膜の優れた側に位置し、内有毛細胞、外有毛細胞の3つの行、および、それぞれの支持細胞の一つの行が含まれています示した。破線1は、コルチ器の解剖のこの臓器中に削除される基底膜に沿って位置を示します。破線2は、マイクロ分離手順の間に除去される基底膜に沿って位置を示します。グリーンはスパイラル輪部の歯間細胞、蝸牛有毛細胞、らせん神経節の神経細胞だけでなく、らせん靱帯と血管条7の細胞を標識するカルビンジンの免疫組織化学的標識を、示している。核のDAPI標識は青色になります。

図2。コルチ器の解剖の臓器。コルティ郭清のハイライトの器官の付属のビデオからの画像)は蝸牛と分離された側頭骨(赤)、B内にある前庭系)蝸牛の骨迷路、C)らせん靱帯と接続されている骨迷路、Dの除去後のコルチ器官)コルティ、Eの臓器かららせん靱帯と血管条(赤)の除去)スパイラル角膜輪部(赤)からの感覚上皮のミクロ分離、およびF)分離されたスパイラル角膜輪部(左)と感覚上皮(右)。

図3。の臓器を示すコルチ植の培養臓器。DIC画像分離されたP4 ATOH1 - nGFPマウスのコルティ、メッキ、と説明したとして、5日間培養した。プロ毛細胞の遺伝子無調相同体1(ATOH1別名Math1を)発現する細胞は、核8にローカライズされている緑色蛍光タンパク質を示すように、このマウスは遺伝的に設計されています。これらのマウスからのコルチ器官は、すべての有毛細胞の核で核GFPのラベルを示すため、落射蛍光顕微鏡を用いて感覚上皮の簡単な可視化を可能にする。比較的大きな渦状角膜輪部は、感覚上皮の外側を見ることができます。コルチ器官から発生した間葉系細胞は、外植片から移行しています。青い核のラベルはDAPIです。
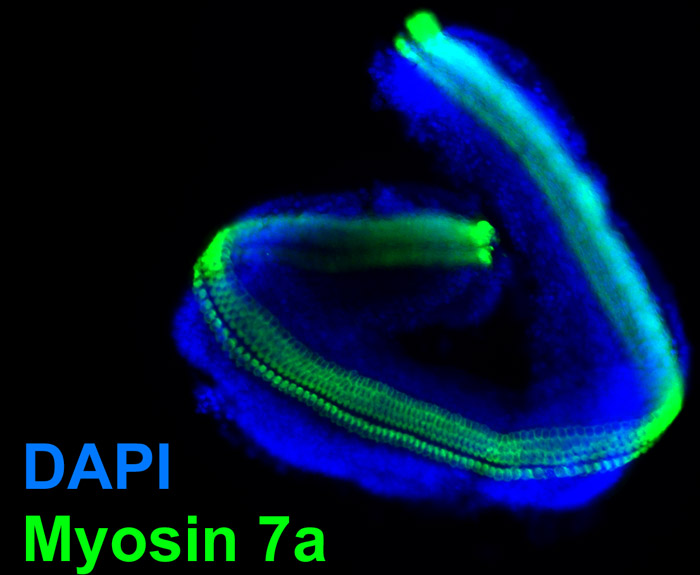
図4。マイクロ分離された感覚上皮に説明し、一晩培養したとしてコルチP4マウス臓器から単離された感覚上皮から得られる。落射蛍光画像。マイクロ分離は、その後4%パラホルムアルデヒドで固定し、蝸牛の有毛細胞を標識するミオシン7aの免疫標識のために処理した。図3から比較的大きな螺旋縁がないことに注意してください。青い核のラベルはDAPIです。

図5。コルチ器の培養臓器にDsRedをレポーター遺伝子のエレクトロポレーション。コルティの全器官がP4 ATOH1 - nGFPの仔マウスから、メッキ、分離して、DsRedをレポーター遺伝子でエレクトロ説明するようにして、落射蛍光顕微鏡下で観察した。トランスジェニックDsRedをレポータータンパク質を発現するコルチ植の臓器の細胞は感覚上皮の赤色蛍光と内因性の細胞を示す緑色の核の蛍光を示す。トランスジェニック細胞は螺旋輪部と感覚上皮を通して見ることができます。
Discussion
この手順を成功させるために重要ないくつかの詳細があります。側頭骨の分離からのコルチ器のインキュベーションの臓器に短い時間、器官は実行可能な器官培養におけるカバースリップし、その結果に添付される可能性が高くなります。したがって、解剖やインキュベーターの器官を置くとの間で時間の量を制限することが重要です。多くのアミノグリコシド系抗生物質が耳に毒性のあるものと有毛細胞の死につながるので、抗生物質の選択は、また非常に重要です。それは完全に抗生物質の使用を見合わせることが望ましいですが、これは汚染の可能性を開いたまま。したがって、我々は潜在的な汚染の問題を克服するための一般的なルールとして10μg/ mLのアンピシリンを使用することを示唆している。
この中で最も厄介な側面、およびコルチ器官の初代培養のための他の手順は、インキュベーションの間にプレートをオフフロートする器官の傾向です。フローティング器官培養は5-7日間実行可能なままことができますが、培養浮遊臓器の欠点があります。例えば、問題のある顕微鏡をレンダリング4-5日後に自分自身の上に頻繁にフォールド器官培養をフローティング。カバースリップに貼付されている器官と比較して、その後、オルガンの構造的完全性が損なわになることができます。私たちは、次のテクニックは、コルチ器官が培地中に浮遊していないことを保証するために役立つことを発見したが、カバースリップに貼付されたままになります。最初に、1:1ポリオルニチン/ラミニンでコートカバーガラスは、前述のように20%FBSを補充した。このプロトコルのために呼ばれる一晩のインキュベーションでは、プレートをコーティングすることを最小の時間です。一週間のための私たちの研究室では、私たちはしばしばコート我々が必要とするプレートのすべてを、それら4℃保管° Cを一日まで、使用前に我々は一晩のインキュベーションのためにインキュベーターに移動したとき。有毛細胞の繊毛が表向きになるように第二に、臓器の後に東洋は、コーティングされたカバーガラスに外植片を輸送される。このオリエンテーションでは、培養皿に基底膜の付着を容易にするでしょう。第三に、外植片は、コートしたディッシュに付するようにうまく内のメディアを削除します。これは、培養皿と基底膜との間の接触を確保し、ガラスに付着する外植片の能力を強化します。これはまた、有毛細胞の行の構造的な整合性を維持する上で支援します。最後に、慎重に200μLの容量のピペットを用いてコルチ器官の表面に培地を2滴を滴下し、その後徐々にカバースリップの側に、残りのボリュームを(合計130μLの)滴下してもして埋める。コーティングされたカバースリップにコルチ器官の添付ファイルを確実に素早く作業することが重要です。外植片のインキュベーションに解剖の開始から、それは通常、コルチ器の分離手順のこの器官を完了するために実践し、オペレータのために10分かかります。
このプロトコルでは、我々はまた、コルチ器官のスパイラル輪部からの感覚上皮の微細分離するための方法を提示する。この手順では、螺旋状角膜輪部は解剖ツールとして28G ½インスリンの注射針を用いて感覚上皮から離れて解剖される。マイクロ分離結果は、有毛細胞とそれらに対応する支持細胞(図3)の行で構成されています。このプロトコルで説明されているように単離した感覚上皮を培養することができます。このマイクロアイソレーション手順は、組織4を周囲からの感覚上皮の酵素の分離と比較する必要があります。哺乳類では、だけでなく、地下間葉系細胞からの鶏、感覚上皮の分離で分離された前庭器官の結果のサーモリシン消化4などの非哺乳類種。ラット蝸牛における、より上皮尾根の分離、低い上皮尾根と基底膜5から付属の感覚上皮におけるサーモリシン消化の結果。しかし、それは蝸牛感覚上皮が付随する間葉系細胞を含まない被覆されたプレートに接続できるかどうかは不明である。機械的なマイクロ分離法とスパイラル角膜輪部からの感覚上皮の分離における酵素消化法の結果、サーモリシン消化上のマイクロ分離解剖の利点の両方が比較的短く、プロトコル、安価な試薬、および潜在的にあまりストレスを含んでいる間消化の酵素の影響による植。また、基底膜は培養プレートへの感覚上皮の付着性を高める可能性がある、このアプローチではそのまま残されます。この方法の欠点は、この繊細な解剖とミクロ解剖から生じる感覚上皮の潜在的な機械的な損傷のためのスキルを開発する必要があります。
この手順のユーティリティの例として、我々はまた、臓器の使用の一例を提示コルティ文化、移植片培養への外来遺伝子のエレクトロポレーション。上記エレクトロポレーション手順は、コルチ器のエレクトロポレーションの臓器の従来の方法に基づいています。特に、鄭と高(2000)植はアガロースの成形溝による電気穿孔のための場所で開催し、無血清培地2のコラーゲンコートした8ウェルLabTekスライド上にしてメッキされているコルチ器の絶縁されたラットの臓器のエレクトロポレーションについて説明します。彼らのアプローチの利点は、臓器が外植片の上面は、理論的には外植片全体でエレクトロポレーションした細胞を均等に分散されるはずカソードを、向くように配向しているということです。コルチ器官、それによりエレクトロポレーション後の外植片の操作を軽減手順を通じてカバーに貼付されて残っているため私たちの手ではしかし、我々が記述するメソッドは、この手順で改善された。さらに、コルチ器官の高い割合は、この提示方法を用いてカバースリップに接続されたままであった。我々の方法は、ジョーンズら (2006)エレクトロポレーション後の遺伝子発現の効率を高めるためのFugene 6試薬の添加を使用してから取得されます。 Jones ら (2006)プロトコルでは、臓器は、エレクトロポレーションされる100 FuGENE 6トランスフェクション試薬のμL、および6メッキで5分間インキュベートした。我々の手法は我々が経験的に最小の臓器毒性との最適なトランスジェニック発現を提供することが見出されたプラスミドDNA、へのFugene 6試薬の3時02分比の使用が異なります。我々は、文化に対する毒性の可能性がある為、原液のFugene 6試薬を使用しないでください。我々のプロトコルにおける電極の構成だけでなく、Jones ら (2006)、螺旋縁または陰極の位置に応じて、感覚上皮のどちらかの主要な遺伝子発現の結果。陰極への遠い文化の側面のDsRedタンパク質陽性細胞があるが、近いカソードへの遺伝子導入細胞の高濃度があります。コルチ植の器官の両側における導入遺伝子の完全な表現を確保するため、現在は単に反転リードして、2番目のパルス列のために逆にすることができます。コルチ器官( 図 5)を通じて導入遺伝子の強い発現に提示プロトコルの結果。
Acknowledgments
著者は、私達にコルチ器官を単離するための方法を教えることへの努力は、DemêmesダニエルとダグラスCotancheに感謝します。とマシューチャナ、ジェイソンミーカー、とケンドラマーシャル(;ビデオアニメーションへの彼女の貢献のアートワークのシェリー林、さらに、我々は、エンジニアリングエレクトロ電極のためにイシュマエルステファノフ-ワーグナーに感謝の意をwww.goodfightproductions.comビデオを生成するため)。 NIDCDからこの作品は、助成金(; RO1DC007174最先端の研究を聴覚のためのP30DC05209 - MEEIのコアサポートR03DC010065 -パーカー)によって賄われていた。
Materials
| Name | Company | Catalog Number | Comments |
| Glass microscope coverslips | DYNALAB Corporation | 2010 | 10mm diameter, circle #1, 1mm thickness, 1 ounce |
| 4 ringed cell culture dish | Greiner Bio-One | 627170 | Sterilized 35 X 10 mm cell culture dish with 4 inner rings |
| Poly-L-ornithine | Sigma-Aldrich | P4957 | 0.01% Solution |
| Laminin | BD Biosciences | 354232 | made in mouse |
| Fetal Bovine Serum | Invitrogen | 26140-095 | Qualified |
| Operating scissors | Roboz Surgical Instruments Co. | RS-6806 | Straight, sharp-blunt length 5" |
| #11 Scalpel Blade | BD Biosciences | 372611 | |
| #4 Dumoxel forceps | Fine Science Tools | 11241-30 | |
| #55 Dumostar fine forceps | Fine Science Tools | 11295-51 | |
| Dulbecco’s Modified Eagle Medium | Invitrogen | 10564-011 | High Glucose |
| Horse Serum | Invitrogen | 2605088 | Heat Inactivated |
| Ampicillin Sodium Salt | Invitrogen | 11593-027 | Irradiated |
| ½ cc Lo-Dose Insulin Syringe | BD Biosciences | 329465 | U-100 28G½ |
| Fugene 6 Transfection Reagent | Roche Group | 11-815-091-001 | |
| Polystyrene test tube | Fisher Scientific | 14-956-5A | |
| Laminar flow hood | The Baker Company (Stanford, ME) | Model SG603a | SterileGARD III Advanced Class II Biological Safety Cabinet |
| Opti-MEM I Reduced-Serum Medium | Invitrogen | 31985 | |
| Reporter plasmid | Clontech Laboratories | 632539 | pCMV DsRed-Express 2 |
| Electroporator | Bio-Rad | 165–2662 | BioRad Gene Pulser Xcell |
References
- Sobkowicz, H. M., Bereman, B., Rose, J. E. Organotypic Development of the Organ of Corti in Culture. Journal of Neurocytology. 119 (4), 543-543 (1975).
- Zheng, J. L., Gao, W. Q. Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nature Neuroscience. 3 (6), 580-580 (2000).
- Martinez-Monedero, R. THE POTENTIAL ROLE OF ENDOGENOUS STEM CELLS IN REGENERATION OF THE INNER EAR. J Neurobiol. 66 (4), 319-319 (2006).
- Saffer, L. D., Gu, R., Corwin, J. T. An RT-PCR analysis of mRNA for growth factor receptors in damaged and control sensory epithelia of rat utricles. Hearing Research. 94 (1,2), 14-14 (1996).
- Zhang, Y. Isolation, growth and differentiation of hair cell progenitors from the newborn rat cochlear greater epithelial ridge. Journal of Neuroscience Methods. 164 (2), 271-271 (2007).
- Jones, J. M. Inhibitors of Differentiation and DNA Binding (Ids) Regulate Math1 and Hair Cell Formation during the Development of the Organ of Corti. J. Neurosci. 26 (2), 550-550 (2006).
- Daniela, B., Josef, S. Calbindin and S100 protein expression in the developing inner ear in mice. The Journal of Comparative Neurology. 513 (5), 469-469 (2009).
- Helms, A. W. Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development. 127 (6), 1185-1185 (2000).
- Zheng, J. L., Wei-Qiang, G. Differential Damage to Auditory Neurons and Hair Cells by Ototoxins and Neuroprotection by Specific Neurotrophins in Rat Cochlear Organotypic Cultures. European Journal of Neuroscience. 8 (9), 1897-1897 (1996).
