Conjugative Mating Assays for Sequence-specific Analysis of Transfer Proteins Involved in Bacterial Conjugation
Summary
Here, we present a protocol to knockout a gene of interest involved in plasmid conjugation and subsequently analyze the impact of its absence using mating assays. The function of the gene is further explored to a specific region of its sequence using deletion or point mutations.
Abstract
The transfer of genetic material by bacterial conjugation is a process that takes place via complexes formed by specific transfer proteins. In Escherichia coli, these transfer proteins make up a DNA transfer machinery known as the mating pair formation, or DNA transfer complex, which facilitates conjugative plasmid transfer. The objective of this paper is to provide a method that can be used to determine the role of a specific transfer protein that is involved in conjugation using a series of deletions and/or point mutations in combination with mating assays. The target gene is knocked out on the conjugative plasmid and is then provided in trans through the use of a small recovery plasmid harboring the target gene. Mutations affecting the target gene on the recovery plasmid can reveal information about functional aspects of the target protein that result in the alteration of mating efficiency of donor cells harboring the mutated gene. Alterations in mating efficiency provide insight into the role and importance of the particular transfer protein, or a region therein, in facilitating conjugative DNA transfer. Coupling this mating assay with detailed three-dimensional structural studies will provide a comprehensive understanding of the function of the conjugative transfer protein as well as provide a means for identifying and characterizing regions of protein-protein interaction.
Introduction
The transfer of genes and proteins at the micro-organismal level plays a central role in bacterial survival and evolution as well as infection processes. The exchange of DNA between bacteria or between a bacterium and a cell can be achieved through transformation, conjugation or vector transduction.1,2 Conjugation is unique in comparison to transformation and transduction in that during conjugation between gram-negative bacteria such as Escherichia coli, the transfer of DNA occurs in a donor-controlled fashion whereby a complex macromolecular system connects donor and recipient cells. Conjugation is also the most direct way in which bacterial cells interact with host cells to inject genes, proteins or chemicals in to host systems.3 Quite often, the transfer of such agents has remarkable effects on the host, ranging from pathogenesis and carcinogenesis to host evolution and adaptation. It has been shown that conjugative recombination increases the rate of adaptation 3-fold in bacteria with high mutation rates under conditions of environmental stress.4 Moreover, conjugation is by far the most common route through which antibiotic resistance genes in bacterial strains are spread.5,6
Microorganisms have evolved specialized secretion systems to support the transfer of macromolecules across cellular membranes; there are currently 9 types of secretion systems (TSSs) in gram negative bacteria that have been described: T1SS, T2SS, T3SS, T4SS, T5SS, T6SS, T7SS, as well as the Sec (secretion) and Tat (two-arginine translocation) pathways.7,8 Each type of secretion system is further divided into different subtypes, a necessity due to diversity of proteins and the distinctiveness of pathways involved, in different bacterial strains. For example, in the type IV secretion system (T4SS), the Ti and Cag systems facilitate effector transport whereas the F-plasmid, R27 and pKM101 T4SSs facilitate transfer of a conjugative plasmid.7,9,10 A detailed understanding of the mechanisms by which organisms assemble their respective secretion systems from their component proteins and share cellular contents with a recipient or their surrounding environment is an important factor in development of targeted strategies to combat pathogenic microorganisms and processes of cellular infection.
Following the initial identification bacterial conjugation in E. coli by Lederberg & Tatum,11 a large number of mobile and conjugative plasmids have been identified and characterized.12 Such mobile plasmids show considerable range is size (from 1 to over 200 kilobases (kb)), however all mobile plasmids contain a relaxase, which recognizes the origin of transfer (oriT) thereby enabling transmission of the plasmid. Conjugative plasmids further encode genes for assembly of a functional T4SS as well as a type IV coupling protein.12 For example the 100 kb F plasmid of E. coli encodes all the conjugative genes within a 33.3 kB transfer (tra) region.13 The genes in the tra region of the F plasmid encode all proteins that facilitate pilus formation, mating pair formation (Mpf), DNA transfer and exclusion functions during conjugative plasmid transfer.10,14,15 A significant body of knowledge is available for conjugative T4SSs, however detailed structural studies of the conjugative proteins and complexes are only more recently becoming available.16–28
In order to assemble a comprehensive view of the conjugative process, a coupling of detailed structural studies to mutational analyses of conjugative transfer proteins is required. This can be achieved through conjugative mating assays. For the F plasmid, each protein encoded within the tra region plays a role in the F-mediated conjugation; therefore, the knockout/deletion of a transfer gene will abolish the conjugative capacity of the cell (Figure 1). While smaller mobile plasmids are more conducive to standard deletion procedures, for larger conjugative plasmids such as F, gene knockouts are more readily achieved via homologous recombination where the target gene is replaced with one conveying a distinct antibiotic resistance gene. In the current protocol, we employ homologous recombination to replace a transfer gene of interest with chloramphenicol acetyltransferase (CAT) in the 55 kb F plasmid derivative pOX38-Tc;29,30 the resultant knockout plasmid, pOX38-Tc Δgene::Cm, facilitates resistance to the presence of chloramphenicol (Cm) in the growth media. Donor cells harboring pOX38-Tc Δgene::Cm are unable to affect conjugative DNA transfer/mating as observed through the use of a mating assay; the mating efficiency of a pOX38-Tc Δgene::Cm donor cell and a normal recipient will decrease or, more often, be abolished. Conjugative transfer of the pOX38-Tc Δgene::Cm plasmid can be restored via a small recovery plasmid harboring the targeted transfer gene. This recovery plasmid can be one that provides constitutive expression, such as plasmid pK184 (pK184-gene),31 or one that provides inducible expression so long as that plasmid properly targets the gene to the correct location within the cell (cytoplasm or periplasm). Consequently, in mating assays between this new donor (harboring pOX38-Tc Δgene::Cm + pK184-gene plasmids) and a recipient cell, the mating efficiency is expected to restore to nearly that of a normal donor-recipient mating assay. This system enables one to probe the function of the knocked out gene through the generation of a series of pK184-gene constructs (deletions or point mutations) and testing each construct's ability to restore the mating capacity of the pOX38-Tc Δgene::Cm harboring donor cells.
Protocol
1. Generation of DNA Constructs
- Designing Oligomers for Homologous Recombination of the Target Gene
- Design a single 55-72 bp forward oligomer as follows: (a) pick a 19-32 bp long nucleotide sequence that is homologous to a DNA sequence in the region 10-100 bp upstream of the 5' start site of the chloramphenicol acetyltransferase gene in the commercial pBAD33 plasmid,32 and (b) select a 36-54 bp long nucleotide sequence homologous to a region 10-150 bp downstream of the 5' start site of the target gene of interest.
- Join the 3' end of nucleotide sequence picked in (b) to the 5' end of the nucleotide sequence picked in (a), thus giving a single 55-72 long forward oligomer.
- Design a single 55-72 bp reverse oligomer as follows: (a) pick a 19-32 bp long nucleotide sequence that is homologous to a DNA sequence in the region 10-100 bp downstream of the 3' end of the chloramphenicol gene in the commercial pBAD33 plasmid, and (b) select a 36-54 bp long nucleotide sequence homologous to a region within 10-150 bp upstream of the 3' end site of the target gene of interest.
- Join the 3' end of nucleotide sequence picked in (b) to the 5' end of the nucleotide sequence picked in (a) to make it a single oligomer.
- Copy this oligomer into any available bioinformatics software and convert sequence into its reverse complement. This will give a single 55-72 bp long reverse oligomer.
- Using any available oligo-analyzer program, check that the recombination oligomers have GC content between 40-60%, low hairpin melting temperature (Tm), low self- and hetero-dimerization Tm's. Order the primers for synthesis and shipment from any preferred biotechnology company.
- Design a single 55-72 bp forward oligomer as follows: (a) pick a 19-32 bp long nucleotide sequence that is homologous to a DNA sequence in the region 10-100 bp upstream of the 5' start site of the chloramphenicol acetyltransferase gene in the commercial pBAD33 plasmid,32 and (b) select a 36-54 bp long nucleotide sequence homologous to a region 10-150 bp downstream of the 5' start site of the target gene of interest.
- Amplification of the CAT Cassette from pBAD33 (CmR) Plasmid Using Oligomers Designed for Homologous Pairing
- Grow an overnight (O/N, 16-18 h) culture of DH5α cells harboring the pBAD33 plasmid in sterile Lysogeny broth (LB) with 20 μg/mL chloramphenicol (Cm) with shaking at 200 rpm and 37 °C.
- Centrifuge 6-8 mL of the O/N culture at room temperature, 5,000 x g for 3 min. Decant the supernatant and extract the pBAD33 plasmid from the pellet using a plasmid mini-prep kit (Materials Table) and manufacturer's protocol.
- Do a digest of the pBAD33 plasmid DNA by adding the following into a sterile tube in the order given: appropriate volume of double distilled water (ddH2O) to a final volume of 50 μL, 1 µg volume of pBAD33 plasmid, 5 μL 10x enzyme reaction buffer, and 0.5 μL of AvaI restriction enzyme (RE). Gently mix by pipetting and let the reaction proceed for 1 h at 37 °C. Heat inactivate the reaction for 20 min (min) at 80 °C. Store samples at -20 °C for no longer than 24 h to minimize sticky-end degradation.
- Prepare a 1.2% agarose separating gel by mixing 1.2 g of agarose with 100 mL of 1x TAE (40 mM Tris, pH 8.5; 20 mM acetic acid; 1 mM EDTA) buffer in a 250 mL Erlenmeyer flask. Heat and swirl to completely dissolve in a microwave. Stop heating immediately when the liquid begins to boil and swirl the flask.
- Cool the liquid agarose for 3 min at room temperature while swirling, add 2 μL of 10 mg/mL ethidium bromide and swirl to mix. Pour the agarose into a gel tray with a well comb and allow it to solidify for 1 h at room temperature. Store gels at 4 °C for up to 2 days in 1x TAE buffer.
- Mix each RE digest from 1.2.3 with 0.2 volume of DNA loading dye (10 μL of dye per 50 μL reaction) by pipetting. Load 5 μL of 500 µg/mL DNA ladder into the first well and all of the RE digest-dye mixture into another well on the agarose gel. Use 2-3 wells to load all of the reaction volume onto the gel.
- Run the gel barely submerged in 1x TAE buffer and operating at 45-50 V (4.5-5 V/cm) for 65 min in a gel electrophoresis device.
- Using a UV cabinet and a sterile razor, quickly cut the 2.8 kb band that corresponds to the CAT sequence out of the gel to minimize UV exposure of DNA. Extract the DNA from the cut-out gel slice using a gel extraction kit (Materials Table) as per the manufacturer's protocol.
Note: Take care not to expose skin directly under UV and handle the razor with care. - Amplify the CAT cassette extracted from 1.2.6 by Polymerase Chain Reaction (PCR) using the primers designed in 1.1 that contain overhangs homologous to the gene sequence to allow for homologous recombination. PCR reactions are set up on ice using the manufacturer guidelines (Materials Table) in the following order:
- Into a sterile PCR tube, add an appropriate volume of ddH2O to a final volume of 50 μL, followed by 10 μL of PCR buffer, 1 μL of 10 mM dNTPs, 2.5 μL of 10 μM Forward primer, 2.5 μL of 10 μM Reverse primer, 1-25 ng of template DNA from 1.2.6 and 0.5 μL of 100 units/μL DNA polymerase.
- Set up a negative control that bears the same components as 1.2.7.1 but with the exception of template DNA. Use an appropriate volume of ddH2O instead of template DNA. Set up a positive control using template DNA and primers that have been proven to work in a PCR reaction, such as those provided as positive control by the manufacturer.
- Mix all reaction contents gently by pipetting.
- Amplify via PCR using the following settings: Initial denaturation for 30 s at 98 °C, 30 cycles of denaturation for 10 s at 98 °C, primer annealing for 20 s, extension for 20 s per kilobase of amplicon at 72 °C and a final extension for 10 min at 72 °C. Store samples at -20 ºC.
- Confirm the correct size of amplification via agarose gel electrophoresis (see 1.2.4-1.2.5) by using only 5 μL of each reaction. Purify the PCR amplicon using a PCR purification kit and manufacturer's protocol. Store purified DNA at -20 ºC.
- The pK184-gene Recovery Plasmid
- Design a forward primer beginning from its 5' end and going towards the 3' end in the following order: (a) pick 4 random nucleotides (a combination of adenine, thymine, guanine and cytosine) for cleavage efficiency, attached to (b) the EcoRI restriction enzyme (RE) cut site sequence (GAATTC), followed by (c) a 21-25 bp long nucleotide sequence that is homologous to the 5' end of the gene of interest, including the start codon. If the target gene contains an EcoRI site, choose another appropriate RE from the pK184 multiple cloning site.
- Design a reverse primer in the following order: (a) pick 4 random nucleotides for cleavage efficiency, followed by (b) a HindIII cut site sequence (AAGCTT) followed by (c) the 21-25 bp long reverse complement of the 3' end of the gene of interest including the stop codon. If the target gene contains a HindIII site, choose another RE in the pK184 multiple cloning site.
- In the case of genes encoding periplasmic proteins, include an additional leader sequence between the RE site and the start codon on the forward primer.
- Using any available oligo-analyzer, check that the primers have GC content between 40-60%, low hairpin Tm, low self- and hetero-dimerization Tm's. Order the primers for synthesis.
- Grow an O/N culture of DY330R pOX38-Tc cells in sterile LB containing 10 μg/mL tetracycline (Tc) with shaking at 32 ºC and 200 rpm. Centrifuge 6-8 mL of the O/N culture at room temperature, 5,000 x g for 3 min. Decant the supernatant and extract the plasmid DNA from the pellet using a plasmid mini-prep kit (Materials Table) and manufacturer's protocol.
- Amplify the full gene of interest with the primers from 1.3.1-1.3.5 using pOX38-Tc as the template (see 1.2.7.1-1.2.7.3).
- Confirm the correct size of amplification via agarose gel electrophoresis (see 1.2.4-1.2.5) by using only 5 μL of each reaction. Purify the amplified DNA using a PCR purification kit and manufacturer's protocol. Store purified DNA at -20 °C.
- Do a double digest of both the commercially available pK184 plasmid DNA and the amplified gene (from 1.3.7.), by adding the following into a sterile tube in the order given: appropriate volume of ddH2O to a final volume of 50 μL, 1 µg volume of pK184 plasmid, 5 μL 10x enzyme reaction buffer, and 1 μL of each EcoRI and HindIII.
- Gently mix by pipetting and let the reaction proceed for 1 h at 37 ºC. Heat inactivate the reaction for 20 min at 80 ºC. Store samples at -20 °C for no longer than 24 h to minimize sticky-end degradation.
Note: The type of restriction nuclease used here depends on the restriction nuclease site that was engineered into the primers in steps 1.3.1 and 1.3.2. - As a positive control, set up single digests of the pK184 plasmid by preparing the same reaction as in 1.3.8 except add only one of the REs to a reaction tube. Do this separately with both REs.
- Gently mix by pipetting and let the reaction proceed for 1 h at 37 ºC. Heat inactivate the reaction for 20 min at 80 ºC. Store samples at -20 °C for no longer than 24 h to minimize sticky-end degradation.
- Run the digests on a 1.2% agarose gel using protocols 1.2.4-1.2.5 Extract the DNA double digest fragments of both pK184 and the gene of interest according to step 1.2.6.
- Ligate the gene insert into the pK184 vector by adding components into a sterile tube in the following order: 2 μL of 10x T4 DNA Ligase Buffer, a total of 100 ng DNA composed of a 1:3 vector:insert (pK184:gene) ratio, ddH2O up to a total volume of 20 μL and 1 μL T4 DNA ligase. As a negative control, set up a similar reaction using 100 ng of vector without the insert gene.
- Gently mix all reaction contents using a micropipette and incubate for 30 min at 25 °C. Heat-inactivate the ligation reaction for 10 min at 65 °C and then place on ice.
- While on ice, add 15 μL of the pK184-gene ligation reaction from step 1.3.10 to 100 μL of chemically competent DH5α cells in a sterile 1.5 mL tube. Gently mix by pipetting and incubate on ice for 10 min. Do the same for the negative control sample. For a positive control, use 20-100 ng of a plasmid such as pBAD33 and transform it into 50 μL of DHα cells.
- Directly transfer the samples from ice into a 42 °C water bath and incubate for 90 seconds. This provides the cells a heat shock and allows them to uptake the plasmid DNA.
- Place cells back on ice for another 5 min and then add 900 μL of sterile LB. Incubate at 37 °C for 1 h while shaking at 125 rpm.
- Aliquot a 100 μL volume of sample from each ligation reaction in 1.3.13 onto an agar plate containing 50 μg/mL kanamycin (Km) and spread the cells using a sterile spreader. The positive control plate should have appropriate antibiotics. Keep the area sterile and work near a flame. Incubate the plate upside down at 37 °C overnight.
- Using a sterile pipette or loop, harvest a single distinct colony of cells and inoculate a 20 mL sterile LB with 50 μg/mL Km. Keep the area sterile and work near a flame. Grow the cells O/N at 37 °C with shaking at 200 rpm.
- Make 3-5 glycerol stocks of the transformed DH5α cells by mixing 500 μL of the O/N culture with 500 μL of sterile 100% glycerol (final 50% v/v) in sterile cryo-tubes. Store at -80 °C.
- Also centrifuge 6-8 mL of the O/N culture at room temperature, 5,000 x g for 3 min. Decant the supernatant and extract the pK184-gene recovery plasmid from the pellet using a plasmid mini-prep kit (Materials Table) and manufacturer's protocol.
- pK184–gene Mutants
Note: Primers designed for deletions, insertions and/or point mutations can be easily generated using manufacturers' online available tools.- Design each forward primer by picking an 18-32 bp long nucleotide sequence that is homologous to the 5' end of the gene of interest, including the start codon. Design primers such that each forward primer anneals 30-180 bp downstream of the preceding one, resulting in deletion mutants lacking N-terminal peptide fragments of appropriate lengths.
- Design a reverse primer by picking an 18-32 bp long nucleotide sequence that is homologous to the 3' end of the gene of interest including the stop codon. Copy this primer into any available bioinformatics program and convert the sequence into its reverse complement. This is the reverse primer.
- In the case of genes encoding periplasmic proteins, design the reverse primer that is the reverse complement of the 3' end of a leader sequence that flanks the 5' end of the gene and is required for proper localization of the protein product in the periplasm.
- Using any available oligo-analyzer program, check that the deletion primers have GC content between 40-60%, low hairpin melting temperature (Tm), low self- and hetero-dimerization Tm's. Order the primers for synthesis and shipment from any available biotechnology company.
- Using the pK184-gene construct obtained in Protocol 1.3 as the template and the guidelines in 1.2.7-1.2.8, PCR amplify the deletion constructs with the primers designed in steps 1.4.1-1.4.3 to generate pK184-geneΔX amplicons. Store amplified DNA at -20 °C.
- Ligate the amplified construct using any available mutagenesis kit (Materials Table).
- Transform each of the pK184-geneΔX ligates separately into chemically competent DH5α cells using a standard heat shock protocol (see 1.3.11-1.3.13).
- Aliquot a 100 μL volume of sample from each ligation reaction in 1.4.12 onto an agar plate containing 50 μg/mL Km and spread the cells using a sterile spreader. Keep the area sterile and work near a flame. Incubate the plate upside down at 37 °C overnight.
- Using a sterile pipette or loop, harvest a single distinct colony of cells and inoculate a 20 mL sterile LB media with 50 μg/mL Km. Keep the area sterile and work near a flame. Grow the cells O/N at 37 °C with shaking at 200 rpm.
- Make 3-5 glycerol stocks of the transformed DH5α cells by mixing 500 μL of the O/N culture with 500 μL of sterile 100% glycerol (final 50% v/v) in sterile cryo-tubes. Store at -80 °C.
- Centrifuge 6-8 mL of the O/N culture at room temperature, 5,000 x g for 3 min. Decant the supernatant and extract the pK184 geneΔX plasmid construct from the pellet using a plasmid mini-prep kit (Materials Table) and manufacturer's protocol. Store DNA at -20 °C.
2. Generation of pOX38-Tc Δgene::Cm Strains
- DY330R pOX38-Tc Δgene::Cm knockouts
- Prepare an O/N culture of DY330R pOX38-Tc cells in 10 mL sterile LB containing 10 µg/mL Tc. Grow culture O/N at 32 °C and 200 rpm.
- Make a 1:70 dilution of the O/N culture into 20 mL of fresh sterile LB. Grow the cells at 32 °C until mid-log phase (OD600nm 0.4-0.6) growth.
- Transfer 10 mL of culture to a sterile flask and incubate at 42 °C for 15 min at 150 rpm in a shaking water bath. This will induce the expression of recombination specific proteins in DY330R.
- Chill the culture in an ice-water bath for 10 min. Prepare electrocompetent cells as follows.
- Transfer the chilled cells into pre-chilled conical tubes and centrifuge at 4,000 x g for 7 min at 4 ºC. All tubes and pipettes to be used in the upcoming steps should be placed at 4 °C or on ice to cool.
- Remove the supernatant and gently resuspended the cells in 1 mL of ice-cold ddH2O. Add another 30 mL of ice cold ddH2O.
- Centrifuge the cells (4,000 x g, 7 min, 4 °C), discard the supernatant and gently resuspended the cells in 1 mL of ice-cold ddH2O.
- Transfer the resuspended cells into pre-chilled 1.5 mL microfuge tubes and centrifuge at 15,000 x g for 1 min at 4 ºC.
- Gently resuspended the pellet in 200 µL of ice-cold ddH2O and aliquot in 50 µL volumes. Electrocompetent cells can be stored at -80 °C
- Add 300 ng of the amplified CAT cassette from 1.2 into 50 μL of electrocompetent DY330R pOX38-Tc cells while mixing on ice, gently by pipetting up and down. Repeat this step using unmodified pBAD33 plasmid as a positive control.
- Transfer the cells to a pre-cooled (-20 °C) 1 mm electroporation cuvette. Electroporate the cells at 1.8 kV with a time constant of 5.5 ms, using an electroporator. Immediately after applying the pulse, dilute the cells with 1 mL of SOC media and transfer to a fresh microfuge tube. Incubate the cells at 32 °C for 2 h.
- Aliquot 100 μL of each sample onto agar plates containing 10 μg/mL Tc and 20 μg/mL Cm and spread using a sterile spreader. Keep the area sterile and work near a flame. Incubate the plate upside down at 32 °C overnight to select for the successful recombinants. The CAT cassette introduced into the cell will undergo homologous recombination with the gene of interest and create the DY330R pOX38-Tc Δgene::Cm (RifR,TcR, CmR) clone.
Note: It is important to grow DY330R cells at 32 °C, with the exception of the 15 min induction at 42 °C of Step 2.1.3 prior to generating electrocompetent cells, as prolonged growth at elevated temperatures risks cell death due to the production of toxic products from the pL operon responsible for recombination functions in DY330R.33,34 - Prepare an O/N of DY330R pOX38-Tc Δgene::Cm cells by harvesting a single distinct colony of cells with a sterile pipette or loop, and inoculating a 20 mL sterile LB media with 10 μg/mL Tc, and 20 μg/mL Cm. Keep the area sterile and work near a flame. Grow the cells O/N at 32 °C with shaking at 200 rpm.
- Make 3-5 glycerol stocks from the O/N by mixing 500 μL of the O/N culture with 500 μL of sterile 100% glycerol (final 50% v/v) in sterile cryo-tubes. Store at -80 °C.
- Centrifuge 6-8 mL of the O/N culture at room temperature, 5,000 x g for 3 min. Decant the supernatant and extract the pOX38-Tc Δgene::Cm construct from the pellet using a plasmid mini-prep kit (Materials Table) and manufacturer's protocol; store purified DNA at -20 °C.
- Perform a conjugative mating assay using XK1200 cells as the recipient to confirm disruption of conjugation by gene knockout as per protocol 3.1.
- DY330R pOX38-Tc Δgene::Cm + pK184-gene
- Transform electrocompetent DY330R pOX38-Tc Δgene::Cm cells with 300 ng of the pK184-gene construct via electroporation as per steps 2.1.4-2.1.7. All selective media must contain 20 μg/mL Cm and 50 μg/mL Km. Incubate at 32 °C. The recovery plasmid in the electroporated cells will now restore the function of the knocked out gene in the DY330R pOX38-Tc Δgene::Cm cells.
- Prepare an O/N of DY330R pOX38-Tc Δgene::Cm + pK184-gene recombinant cells by harvesting a single distinct colony of the cells with a sterile pipette or loop, and inoculating a 20 mL sterile LB media with 20 μg/mL Cm and 50 μg/mL Km. Keep the area sterile and work near a flame. Grow the cells O/N at 32 °C with shaking at 200 rpm.
- Make 3-5 glycerol stocks from the O/N by mixing 500 μL of the O/N culture with 500 μL of sterile 100% glycerol (final 50% v/v) in sterile cryo-tubes. Store at -80 °C.
- Also perform protocol 3.1 to generate XK1200 pOX38-Tc Δgene::Cm recombinant cells.
- XK1200 pOX38-Tc Δgene::Cm + pK184-gene and Mutants
- Prepare electrocompetent XK1200 pOX38-Tc Δgene::Cm cells (from step 2.2.4).
- Separately electroporate 300 ng of pK184-gene or pK184-gene mutant plasmids into 50 μL of electrocompetent XK1200 pOX38-Tc Δgene::Cm cells as per steps 2.1.4-2.1.7. All selective media should contain 10 μg/mL nalidixic acid (Nal), 20 μg/mL Cm, and 50 μg/mL Km and be incubated at 37 °C.
- Prepare an O/N of XK1200 pOX38-Tc Δgene::Cm+ pK184-gene recombinant cells by harvesting a single distinct colony of the cells with a sterile pipette or loop, and inoculating a 20 mL sterile LB media with Nal, 20 μg/mL Cm and 50 μg/mL Km. Keep the area sterile and work near a flame. Grow the cells O/N at 37 °C with shaking at 200 rpm.
- Make 3-5 glycerol stocks from the O/N by mixing 500 μL of the O/N culture with 500 μL of sterile 100% glycerol (final 50% v/v) in sterile cryo-tubes. Store at -80 °C.
3. Conjugative Mating Assays
- Conjugative Mating to Generate XK1200 pOX38-Tc Δgene::Cm Cells
- Prepare a 20 mL sterile LB O/N culture of DY330R pOX38-Tc Δgene::Cm + pK184-gene cells by using a sterile pipette or loop to inoculate a 20 mL sterile LB containing 20 μg/mL Cm and 50 μg/mL Km with a glycerol stock or single colony on an agar plate. Grow at 32 °C with shaking at 200 rpm. Prepare the same for XK1200 cells in 20 mL sterile LB with 10 μg/mL Nal, growing at 37 °C.
- Make 1:70 dilutions of each culture separately in 2 mL of sterile LB with the same antibiotic contents; add glucose to a final concentration of 100 mM to all donor cells. Grow cells to mid-log phase (OD600 0.5-0.7) at 37 °C with shaking at 200 rpm.
- Centrifuge (4,000 x g for 5 min at 4 °C) to pellet cells, discard supernatant, wash once with cold sterile LB to remove antibiotics, and resuspend cells in 2 mL cold sterile LB.
- Aliquot 100 μL of each culture into 800 μL of sterile LB media and allow them to mate at 32 °C for 1 h without shaking.
- Vortex the cells for 30 s to disrupt the mating pairs and place them on ice for 10 min to prevent further mating.
- Aliquot 100 μL of the cell mixture onto an agar plate containing 10 μg/mL Nal and 20 μg/mL Cm to select for XK1200 pOX38-Tc Δgene::Cm cells. Spread the cells using a sterile spreader. Keep the area sterile and work near a flame. Incubated the plate O/N at 37 °C upside-down.
- Harvest a single colony of the new XK1200 pOX38-Tc Δgene::Cm knockout strain using a sterile pipette or loop and grow in sterile LB with 20 μg/mL Cm, O/N at 37 °C with shaking at 200 rpm. Make 3-5 glycerol stocks from the O/N by mixing 500 μL of the O/N culture with 500 μL of sterile 100% glycerol (final 50% v/v) in sterile cryo-tubes. Store at -80 °C.
Note: The resultant cells are now able to be made competent for transformation with the pK184-gene constructs (protocols 1.3 and 1.4) for assessing the gene and its mutants on the ability to recover conjugative transfer in protocol 3.2.
- Conjugative Mating Assay from XK1200 Donors to MC4100 Recipients
- Prepare an O/N culture of XK1200 pOX38-Tc Δgene::Cm + pK184-gene cells in 20 mL of sterile LB with 20 μg/mL Cm, 50 μg/mL Km and MC4100 cells in 5 mL LB with 50 μg/mL streptomycin (Sm) using cells from a glycerol stock or single colony on an agar plate and sterile pipette or loop. Grow cultures at 37 °C with 200 rpm shaking.
- Make 1:70 dilutions from each O/N culture separately in 2 mL of sterile LB with the same antibiotics. Add glucose to a final concentration of 100 mM to all donor cells. Grow cells to mid-log phase (OD600 0.5-0.7) at 37 °C with shaking at 200 rpm.
- Centrifuge (4,000 x g for 5 min at 4 °C) to pellet cells, discard supernatant, wash once with cold sterile LB to remove antibiotics, and resuspend cells in 2 mL cold sterile LB.
- In duplicate, aliquot 100 μL of each culture into 800 μL of sterile LB media and allow them to mate at 37 °C for 1 h without shaking.
- Vortex the cells for 30 s to disrupt the mating pairs and place them on ice for 10 min to prevent further mating.
- Using the mid-log cultures from step 3.2.2 and fresh sterile LB, prepare 6 serial dilutions of the donor and recipient cells from 10-2 to 10-7.
- On each of two halves of an agar plate containing 10 μg/mL Nal, 20 μg/mL Cm and 50 μg/mL Km, spot 10 μL aliquots of each dilution of XK1200 donor cells, as shown in Figure 2. Repeat for the dilutions of the recipient MC4100 cells on agar plates containing 50 μg/mL Sm. Incubate plates O/N at 37 °C.
- Using the vortexed mixture from step 3.2.5 and fresh sterile LB, prepare 6 dilutions (10-2 to 10-7) of the transconjugants. Select for the transconjugant MC4100 pOX38-Tc Δgene::Cm cells by spotting 10 μL aliquots of each of dilution on each half of agar plates containing 50 μg/mL Sm and 20 μg/mL Cm, as in Figure 2. Repeat for both duplicate mixtures. Keep the area sterile and work near a flame. Incubate plates O/N at 37 °C.
- Determine the mating efficiency of each construct as described in protocol 3.3.
- Repeat this protocol for all recovery plasmids to evaluate the effect of a particular mutation on the efficiency of conjugation.
- Calculation of Mating Efficiency
- Count the number of colonies from the same dilution spotting for each donor, recipient and transconjugant cells on their respective plates.
- Count recipient colonies to test any bias that would result from having a larger number of transconjugants than recipients at that given dilution.
- Calculate the mating efficiency as the number of transconjugant colonies divided by number of donor colonies. Multiply by 100 to obtain efficiency value per 100 donor cells.
Representative Results
The process of F plasmid-driven bacterial conjugation is a coordinated process that involves transfer proteins within the tra region of the F-plasmid that assembles a T4SS to facilitate pilus synthesis and conjugative DNA transfer. The protein TraF (GenBank accession # BAA97961; UniProt ID P14497) is required for conjugative F-pilus formation.10,14,35–37 The protein contains a C-terminal thioredoxin-like domain, though it does not have the catalytic CXXC motif.35,38 Although it has been predicted to interact with the TraH protein through its N-terminal domain,39 a region shown to be more dynamic than its C-terminal domain,37 not much else is known about the protein's structural features. To assess the functional aspects of the TraF protein in conjunction with structural studies, we first knocked out the traF gene on the pOX38-Tc via homologous recombination, generating the pOX38-Tc ΔtraF::Cm plasmid (Table 1) in E. coli DY330R cells.33,34 Also generated was the pK184-TraF plasmid from traF-specific primers (Table 2) to provide recovery of conjugation and enable probing the protein's sequence. The transfer of the pOX38-Tc ΔtraF::Cm plasmid into XK1200 cells40 from DY330R cells when pK184-TraF was provided in trans (transconjugants grown on 10 μg/mL Nal, 10 μg/mL Tc and 20 μg/mL Cm) indicates that (a) the traF knockout in pOx38-Tc ΔtraF::Cm provides an in-frame CAT cassette, and (b) that pK184-TraF can restore conjugative function.
A series of TraF mutants were generated for analysis using the conjugative mating assay37 using XK1200 pOX38-Tc ΔtraF::Cm + pK184-TraFΔX cells and MC4100 cells41 as donors and recipients, respectively. One representative mutant is TraF55-247 (Table 1), an N-terminal deletion mutant that removes the region of the protein predicted to interact with TraH. When the full-length TraF protein is provided to the donor cells, conjugative transfer is restored (Figure 2), while providing the empty plasmid pK184 does not (Table 3). Similarly, conjugative function in the XK1200 donor cells is not restored when provided with plasmid pK184-TraF55-247 (Table 3). This indicates that the truncated region of the protein is important for TraF's function within the conjugative apparatus, likely through interaction with TraH, and provides a region of the protein to target for further mutational analysis.
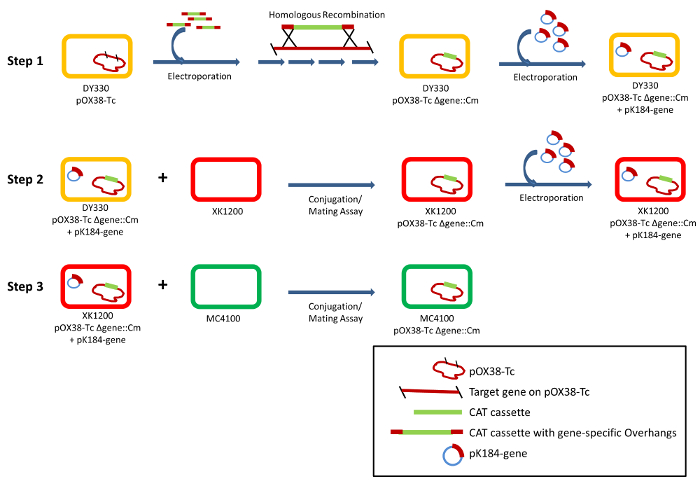
Figure 1: Schematic representation of the conjugative mating assay. In Step 1, target genes are knocked out by homologous recombination in DY330R cells, an efficient recombination strain, using a PCR generated CAT cassette with overhangs homologous for the target gene. The resultant DY330R clone harbors the plasmid pOX38-Tc Δgene::Cm, which is unable to facilitate conjugation unless the knocked out gene is provided in trans via a recovery plasmid (pK184-gene). The resultant pOX38-Tc Δgene::Cm plasmid is transferred to a XK1200 strain (Step 2) for further assessment of the gene using mating assays with a MC4100 recipient (Step 3). Please click here to view a larger version of this figure.
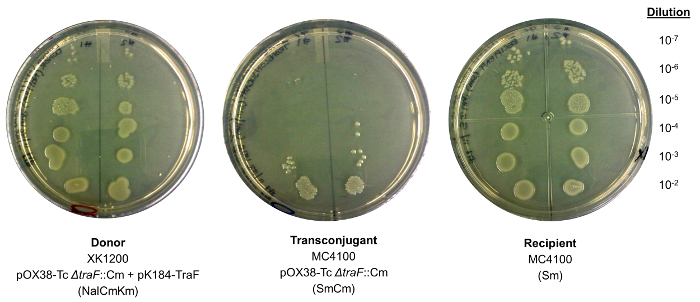
Figure 2: Mating assay to assess the target gene's function in conjugation. Donor and recipient cells were E. coli XK1200 pOX38-Tc ΔtraF::Cm transformed with pK184-TraF and MC4100, respectively. The resulting transconjugants (MC4100 pOX38-Tc ΔtraF::Cm) grow on plates containing Sm and Cm, indicating successful recovery of conjugative function. The experiment is done in duplicate on a single agar plate, and serial dilutions are used in order calculate the mating efficiency of restorative gene mutants (Table 2). Please click here to view a larger version of this figure.
| Bacterial strain/Plasmid | Relevant Characteristics | Selective Marker(s)† | Reference |
| Bacterial Strains | |||
| DY330R | W3110 ΔlacU169 gal490 λc1857 Δ(cro-bioA) RifR | Rif | 33,34 |
| XK1200 | F– lacΔU124 Δ(nadA aroG gal attλ bio gyrA) | Nal | 40 |
| MC4100 | araD139 Δ(argF-lac)U169 rpsL150 relA1 flbB3501 deoC1 ptsF25 rbsR | Sm | 41 |
| Vectors and Constructs | |||
| pBAD33 | Plasmid for expression under from ParaBAD | Cm | 32 |
| pK184 | 2.4 kb cloning vector, p15a replicon | Km | 31 |
| pK184-TraF‡ | F TraF from pOX38 in pK184 | Km | 35 |
| pK184-TraF56-247 | F TraF aa 56-247 from pK184-TraF | Km | |
| Conjugative Plasmids | |||
| pOX38-Tc | IncFI, Tra+, RepFIA+, f1 HindIII fragment of F, mini-Tn | Tc | 29,30 |
| pOX38-Tc ΔtraF::Cm | pOX38-Tc with CAT inserted in traF | Tc Cm | 35 |
| †Cm, chloramphenicol; Km, kanamycin; Nal, nalidixic acid; Rif, rifampicin; Sm, streptomycin; Tc, tetracycline | |||
| ‡All TraF constructs contain the 19-residue leader sequence to ensure localization to the periplasmic space. | |||
Table 1: E. coli strains and plasmids used in this study.
| Construct | Primer* |
| TraF-Cm-For | 5’- GATCGAGGCTGGCAGTGGTATAACGAGAAAATAAATCCGAAGGA – CTGTGACG GAAGATCACTTC -3’ |
| TraF-Cm-Rev | 5’- TCTTCAGAAACGTTCAGGAACTGTTTTGCCAGGTCGTCCT – CTTATTCAGGCGT AGCACCAG -3’ |
| pK184-TraF-For | 5’- TTTTTTGAATTCTATGAATAAAGCATTACTGCCAC -3’ |
| pK184-TraF-Rev | 5’- TTTTTTAAGCTTTAAAAATTGGGTTTAAAATCTTCAGAAA -3’ |
| pK184-TraF55-247-For | 5’- TACGCATATGATGGCCGCACTGCAGACGG -3’ |
| pK184-TraF55-247-Rev | 5’- TACGCATATGTCCTGACGCCGGAAAAATAAAGCAGCAGAGTAA -3’ |
| †Table adapted from Lento et al. 201635 with permission | |
| *TraF overhanging regions are italicized. Restriction enzyme sites are underlined (HindIII: AAGCTT, EcoRI: GAATTC, NdeI: CATATG) | |
Table 2: A list of primers used in this study.
| Donor Plasmid† | Recovery Plasmid | Transconjugants‡§ (cells mL-1) | Mating Efficiency§|| |
| pOX38-Tc ΔtraF::Cm | None | 0 | 0 |
| pOX38-Tc ΔtraF::Cm | pK184 | 0 | 0 |
| pOX38-Tc ΔtraF::Cm | pK184-TraF | 5 x 103 | 0.0167 |
| pOX38-Tc ΔtraF::Cm | pK184-TraF55-247 | 0 | 0 |
| *Table adapted from Lento et al. 201635 with permission | |||
| †Donor cells were E. coli XK1200, with an average concentration of 3 x 107 cells mL-1 | |||
| ‡Recipient cells were E. coli MC4100. The number of transconjugants for the positive control is from a 10-5 dilution. 0 indicates no transconjugants from a 10-2 dilution. | |||
| §An average of two to four mating experiments were performed for each construct. | |||
| ||Mating efficiency is defined as transconjugants per 100 donor cells. 0 mating efficiency indicates no transconjugants from a 10-2 dilution | |||
Table 3: Abolished mating efficiency by TraF deletion constructs.
Discussion
Bacterial conjugation process provides a means by which bacteria can spread genes providing an evolutionary advantage for growth in challenging environments, such as the spread of antibiotic resistance markers. Because many of the conjugative plasmids are so large,12 functional studies on the proteins involved in assembly of the transfer apparatus through mutation of target genes on the conjugative plasmid itself are unwieldy. The protocols detailed herein provide a means by which one can more readily assess the target gene of interest through the use of smaller, more manageable expression plasmids (Figure 1). We employ the F plasmid derivative pOX38-Tc (Table 1) to study F plasmid-mediated conjugation; other conjugative plasmids can be studied using the protocols detailed here and appropriate derivative plasmids. The mating assays outlined have been adapted from Frost and colleagues,42 with some modifications. In previous studies,8,33,42 the creation of the pOX38-Tc Δgene::Cm construct was achieved by cleaving the gene of interest with the appropriate restriction enzymes and inserting the amplified CAT cassette into pOX38-Tc.42 In the current method, we employ homologous recombination in the recombineering E. coli strain DY330R33,34 to knockout the target gene and replace it with the CAT cassette. This has an advantage of allowing the resultant DY330R strain harboring the pOX38-Tc Δgene::Cm construct to act as a control for the gene-specific knockout out F-T4SS mediated conjugative transfer via the recovery of transfer using the pK184-gene recovery plasmid. While it may be possible to generate knockouts using a CRISPER-Cas9 methodology,43 we have not at this time explored this possibility.
The process begins with the generation of a gene knockout in the F derivative plasmid pOX38-Tc (Figure 1). This is achieved via homologous recombination in DY330R cells (Table 1), other strains with similar features such as DY329, DY331 and DY378 can also be used. Primers are initially designed to PCR amplify the CAT cassette from the pBAD33 plasmid32 and contain overhanging bases that are specific for the target gene (Table 2); the PCR product is then electroporated into DY330R cells harboring pOX38-Tc. Homologous recombination generates the knock-out plasmid pOX38-Tc Δgene::Cm where the CAT cassette is inserted in-frame within the target gene, effectively disrupting T4SS assembly and conjugation while providing Cm resistance. At the same time, the target gene is PCR amplified from pOX38-Tc and inserted into a small expression plasmid; in this protocol we use pK184 for constitutive expression, however one could chose a plasmid with inducible expression of the target gene if desired. The pK184-gene plasmid is then transformed into the DY330R pOX38-Tc Δgene::Cm cells, and these cells are then used as donors to transfer the pOX38-Tc Δgene::Cm construct via conjugation into XK1200 cells for mating assays. In the mating assays, the donor and recipient cells are XK1200 pOX38-Tc Δgene::Cm and MC4100, respectively. The pK184-gene recovery plasmid, as well as the series mutants of the target gene (deletions or point mutations), is provided to the donor cells to assess their ability to restore mating, and determine its efficiency, with MC4100 recipient cells (Figure 2; Table 3).
Critical to the procedure is the use of appropriate donor and recipient strains (Table 1), and the design of the primers used. For each primer designed, there are general guidelines that should be followed. While we strictly try to abide with 40-60% GC content, this may not always be possible. In such cases, it is the experimenter's discretion to test a primer with GC content slightly below or above this range. The melting temperature (Tm) of the forward and reverse primer must be similar, and the annealing temperature (Ta) value should always be lower than the Tm by 2-5 °C for PCR. The free energy available for allowing a hairpin to develop should be much lower than the Ta, while the homo- and hetero- dimerization Tm's must be very low (less than -10 kJ and 30 °C). Primers can be designed to probe deletions, insertions or point mutations as desired. It is of course critical that the resultant gene construct be amplified and ligated into the recovery plasmid in-frame such that it is properly expressed. Transformation of competent cells can be done via electroporation or heat shock using electrocompetent or chemically competent cells, respectively. We find that electroporation is more efficient for larger constructs such as pOX38-Tc and the homologous recombination oligonucleotide, while the smaller expression plasmids such as pK184-TraF can be readily transformed into cells using heat shock methods. Lastly, it is important to remember that there will be multiple antibiotics in use throughout the protocol, as both donor and recipient strains require different resistances that are different from the ones employed on the conjugative and recovery plasmids.
Aside from the mating assays techniques described here, there are other methods that are used to study bacterial conjugation, varying slightly in their approach. Horizontal gene transfer is a process where bacteria transfer a plasmid to a recipient cell, including interspecies recipients.44 A study by Dahlberg and colleagues44 for instance used bacterial conjugation to determine the extent of interspecies horizontal gene transfer. They utilized the incorporation of green fluorescent protein (GFP) into a plasmid cloned into KT2442 cells; the chromosomal lac Iq gene in the KT2442 cells repress GFP expression. When the plasmid carrying GFP is transferred to a species without the lac Iq gene, fluorescence is observed.44 Despite its limitation to provide protein specific function in various species, the interspecies conjugation experiment could possibly be coupled with the protocols presented here to make evolutionary predictions for protein-protein interactions between different species.
Declarações
The authors have nothing to disclose.
Acknowledgements
This research was supported by a Discovery Grant from the Natural Sciences & Engineering Council of Canada (NSERC).
Materials
| GeneJet Plasmid Mini-Prep Kit | Fisher Scientific | K0503 | |
| GeneJet Gel Extraction Kit | Fisher Scientific | K0692 | |
| GeneJet PCR Purification Kit | Fisher Scientific | K0702 | |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | E0554S | |
| Broad Range DNA Ladder | New England Biolabs | N0303A | |
| Petri Dishes | Fisher Scientific | FB0875713 | |
| Electroporator | Eppendorf | 4309000027 | |
| Electroporation cuvettes | Fisher Scientific | FB101 | Cuvettes have a 1 mm gap. |
| Enzymes | |||
| AvaI | New England Biolabs | R0152S | |
| EcoRI | New England Biolabs | R0101S | |
| HindIII | New England Biolabs | R0104L | |
| NdeI | New England Biolabs | R0111S | |
| Phusion DNA Polymerase | New England Biolabs | M0530L | |
| T4 DNA Ligase | New England Biolabs | M0202S | |
| DpnI | New England Biolabs | R0176S | |
| Antibiotics | Final Concentrations | ||
| Chloramphenicol (Cm) | Fisher Scientific | BP904-100 | 20 µg/mL |
| Kanamycin (Km) | BioBasic Inc. | DB0286 | 50 µg/mL |
| Nalidixic acid (Nal) | Sigma-Aldrich | N8878-25G | 10 µg/mL |
| Rifampicin (Rif) | Calbiochem | 557303 | 20 µg/mL |
| Tetracycline (Tc) | Fisher Scientific | BP912-100 | 10 µg/mL |
| Streptomycin (Sm) | Fisher Scientific | BP910-50 | 50 µg/mL |
Referências
- Dobrindt, U., Hochhut, B., Hentschel, U., Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol. 2 (5), 414-424 (2004).
- Furuya, E. Y., Lowy, F. D. Antimicrobial-resistant bacteria in the community setting. Nat Rev Microbiol. 4 (1), 36-45 (2006).
- Griffiths, A., Miller, J., Suzuki, D., Lewontin, R., Gelbart, W. . An Introduction to Genetic Analysis, 7th Edition. , (2000).
- Cooper, T. F., Barton, N. H. Recombination Speeds Adaptation by Reducing Competition between Beneficial Mutations in Populations of Escherichia coli. PLoS Biol. 5 (9), 225 (2007).
- Lujan, S. A., Guogas, L. M., Ragonese, H., Matson, S. W., Redinbo, M. R. Disrupting antibiotic resistance propagation by inhibiting the conjugative DNA relaxase. Proc Natl Acad Sci. 104 (30), 12282-12287 (2007).
- Carattoli, A. Plasmids and the spread of resistance. Int J Med Microbiol. 303 (6-7), 298-304 (2013).
- Shala, A., Ferraro, M., Audette, G. F., Bawa, R., Audette, G. F., Rubenstein, I. Bacterial Secretion Systems: Nanomachines for Infection and Genetic Diversity. Handbook of Clinical Nanomedicine: Nanoparticles, Imaging, Therapy and Clinical Applications. , 657-686 (2016).
- Tseng, T. T., Tyler, B. M., Setubal, J. C. Protein secretion systems in bacterial-host associations, and their description in the Gene Ontology. BMC Microbiol. 9 (1), 2 (2009).
- Alvarez-Martinez, C. E., Christie, P. J. Biological diversity of prokaryotic type IV secretion systems. Microbiol Mol Biol Rev. 73 (4), 775-808 (2009).
- Lawley, T. D., Klimke, W. A., Gubbins, M. J., Frost, L. S. F factor conjugation is a true type IV secretion system. FEMS Microbiol Lett. 224 (1), 1-15 (2003).
- Lederberg, J., Tatum, E. L. Gene Recombination in Escherichia Coli. Nature. 158 (4016), 558-558 (1946).
- Smillie, C., Garcillan-Barcia, M. P., Francia, M. V., Rocha, E. P. C., de la Cruz, F. Mobility of Plasmids. Microbiol Mol Biol Rev. 74 (3), 434-452 (2010).
- Willetts, N., Skurray, R. The Conjugation System of F-Like Plasmids. Annu Rev Genet. 14 (1), 41-76 (1980).
- Frost, L. S., Ippen-Ihler, K., Skurray, R. A. Analysis of the sequence and gene products of the transfer region of the F sex factor. Microbiol Rev. 58 (2), 162-210 (1994).
- Audette, G. F., Manchak, J., Beatty, P., Klimke, W. A., Frost, L. S. Entry exclusion in F-like plasmids requires intact TraG in the donor that recognizes its cognate TraS in the recipient. Microbiology. 153, 442-451 (2007).
- Christie, P. J., Atmakuri, K., Krishnamoorthy, V., Jakubowski, S., Cascales, E. Biogenesis, Architecture, and Function of Bacterial Type Iv Secretion Systems. Annu Rev Microbiol. 59 (1), 451-485 (2005).
- Christie, P. J. Type IV secretion: the Agrobacterium VirB/D4 and related conjugation systems. Biochim Biophys Acta – Mol Cell Res. 1694 (1-3), 219-234 (2004).
- Bhatty, M., Laverde Gomez, J. a., Christie, P. J. The expanding bacterial type IV secretion lexicon. Res Microbiol. 164 (6), 620-639 (2013).
- Christie, P. J., Cascales, E. Structural and dynamic properties of bacterial Type IV secretion systems (Review). Mol Membr Biol. 22 (1-2), 51-61 (2005).
- Christie, P. J. Type IV secretion: Intercellular transfer of macromolecules by systems ancestrally related to conjugation machines. Mol Microbiol. 40 (2), 294-305 (2001).
- Christie, P. J., Whitaker, N., González-Rivera, C. Mechanism and structure of the bacterial type IV secretion systems. Biochim Biophys Acta. 1843 (8), 1578-1591 (2014).
- Cascales, E. The type VI secretion toolkit. EMBO Rep. 9, 735 (2008).
- Silverman, J. M., Brunet, Y. R., Cascales, E., Mougous, J. D. Structure and Regulation of the Type VI Secretion System. Annu Rev Microbiol. 66 (1), 453-472 (2012).
- Chandran, V., et al. Structure of the outer membrane complex of a type IV secretion system. Nature. 462 (7276), 1011-1015 (2009).
- Rivera-Calzada, A., et al. Structure of a bacterial type IV secretion core complex at subnanometre resolution. EMBO J. 32 (8), 1195-1204 (2013).
- Waksman, G., Fronzes, R. Molecular architecture of bacterial type IV secretion systems. Trends Biochem Sci. 35, 691 (2010).
- Fronzes, R., Christie, P. J., Waksman, G. The structural biology of type IV secretion systems. Nat Rev Microbiol. 7 (10), 703-714 (2009).
- Kaplan, M., et al. Probing a cell-embedded megadalton protein complex by DNP-supported solid-state NMR. Nat Methods. 12 (7), 5-9 (2015).
- Guyer, M. S., Reed, R. R., Steitz, J. A., Low, K. B. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harb Symp Quant Biol. 45, 135-140 (1981).
- Anthony, K. G., Sherburne, C., Sherburne, R., Frost, L. S. The role of the pilus in recipient cell recognition during bacterial conjugation mediated by F-like plasmids. Mol Microbiol. 13 (6), 939-953 (1994).
- Jobling, M. G., Holmes, R. K. Construction of vectors with the pl5a replicon, kanamycin resistance, inducible lacZα and pUC18 or pUC19 multiple cloning sites. Nucleic Acids Res. 18 (17), 5315 (1990).
- Guzman, L. M., Belin, D., Carson, M. J., Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose P(BAD) promoter. J Bacteriol. 177 (14), 4121-4130 (1995).
- Yu, D., et al. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A. 97 (11), 5978-5983 (2009).
- Lawley, T. D., Gilmour, M. W., Gunton, J. E., Standeven, L. J., Taylor, D. E. Functional and Mutational Analysis of Conjugative Transfer Region 1 (Tra1) from the IncHI1 Plasmid R27. J Bacteriol. 184 (8), 2173-2180 (2002).
- Elton, T. C., Holland, S. J., Frost, L. S., Hazes, B. F-Like Type IV Secretion Systems Encode Proteins with Thioredoxin Folds That Are Putative DsbC Homologues. J Bacteriol. 187 (24), 8267-8277 (2005).
- Hazes, B., Frost, L. Towards a systems biology approach to study type II/IV secretion systems. Biochim Biophys Acta. 1778, 1839-1850 (2008).
- Lento, C., Ferraro, M., Wilson, D., Audette, G. F., Tsolis, R. HDX-MS and deletion analysis of the type 4 secretion system protein TraF from the Escherichia coli F plasmid. FEBS Lett. 590 (3), 376-386 (2016).
- Audette, G. F., Van Schaik, E. J., Hazes, B., Irvin, R. T. DNA-binding protein nanotubes: Learning from nature’s nanotech examples. Nano Lett. 4, 1897-1902 (2004).
- Harris, R. L., Silverman, P. A. Tra proteins characteristic of F-like type IV secretion systems constitute an interaction group by yeast two-hybrid analysis. J Bacteriol. 186 (16), 5480-5485 (2004).
- Moore, D., et al. Characterization of the F-Plasmid Conjugative Transfer Gene traU. J Bacteriol. 172 (8), 4263-4270 (1990).
- Anthony, K. G., Sherburne, C., Sherburne, R., Frost, L. S. The role of the pilus in recipient cell recognition during bacterial conjugation mediated by F-like plasmids. Mol Microbiol. 13 (6), 939-953 (1994).
- Klimke, W. A., Frost, L. S. Genetic analysis of the role of the transfer gene, traN, of the F and R100-1 plasmids in mating pair stabilization during conjugation. J Bacteriol. 180 (16), 4036-4043 (1998).
- Jiang, W., Marraffini, L. A. CRISPR-Cas: New Tools for Genetic Manipulations from Bacterial Immunity Systems. Annu Rev Microbiol. 69 (1), 209-228 (2015).
- Dahlberg, C., Bergstrom, M., Andreasen, M., Christensen, B. B., Molin, S., Hermansson, M. Interspecies bacterial conjugation by plasmids from marine environments visualized by gfp expression. Mol Biol Evol. 15 (4), 385-390 (1998).
