Small rings consisting of cyclopropanes, oxiranes, and aziridines are found in various compounds such as natural products and drugs1,2. They are primarily used as starting materials exploiting their ring strain. Among the three-ring compounds, aziridine has been studied less extensively due to its instability and uncontrollable reactivity3. As shown in the electrostatic potential maps (Figure 1), a group attached to the aziridine ring-nitrogen, whether electron-donating or electron-attracting, makes the basicity of nitrogen different. This difference provides a striking contrast to the reactivity and selectivity of the corresponding aziridines.
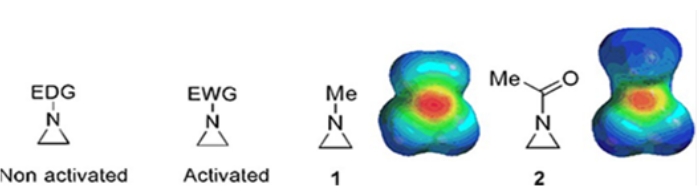
Figure 1: Chemical structures of "activated" and "non-activated" aziridines and electrostatic potential maps of their representative examples N-methylaziridine, and N-acetylaziridine4. This figure has been modified with permission from Ranjith et al.4. Please click here to view a larger version of this figure.
When the ring nitrogen has an electron-withdrawing group, such as sulfonate, phosphonate, and carbamate, we call it "activated" aziridine. This is readily reactive with nucleophiles to compensate for its instability with a limited scope of regiochemistry. These activated aziridines are prepared through various catalytic methods and used as a starting material. Much of recent aziridine chemistry has dealt with these activated aziridines. However, activated aziridines suffer certain restrictions resulting from their instability and limited reaction scope of the ring opening. On the other hand, aziridines bearing electron-donating substituents, like alkyl or substituted alkyl groups, at the ring nitrogen called "non-activated"4, are relatively stable under most circumstances and can be left on the bench for a long time without significant decomposition. The nucleophilic ring-opening reactions of non-activated aziridine occur via the formation of aziridinium ions. Most reactions of aziridine ring-opening and ring transformations proceed in a highly regiochemical manner. However, very few literature reports discuss the preparation of optically pure non-activated aziridines with substituents at the C2 or C3 positions5,6.
This paper shows the successful preparation of α-methylbenzyl group-containing chiral aziridine-2-carboxylate derivatives, specifically (-)-mentholyl (1R)-phenylethylaziridine-2-carboxylates as its diastereomeric mixture, from the reaction of 2,3-dibromopropionate and (1R)-phenylethylamine. From this diastereomeric mixture, enantiopure (1R)-phenylethyl-(2R)- and (2S)-aziridine-2-carboxylates as their (-)-mentholyl esters were obtained in optically pure forms by selective recrystallization from MeOH and n-pentane on multi-hundred-kilo scales (Figure 1)7. These (-)-mentholyl esters can be easily converted into their ethyl or methyl esters by transesterification in the presence of magnesium or potassium carbonate7. These compounds can also be prepared easily on a laboratory scale from the reactions of alkyl 2,3-dibromopropionates or the vinyl triflate of α-ketoester with chiral 2-phenylethylamine followed by separation of the diastereomeric mixture using simple flash column chromatography8.
Once we have enantiopure chiral aziridine-2-carboxylate, we can synthesize various cyclic and acyclic nitrogen-containing biologically important target molecules based on functional group transformations of carboxylate and highly regio- and stereoselective aziridine-ring opening reactions6,9,10. The first expedient asymmetric synthesis was applied for both enantiomers of 5, 6-dihydrouracil-type marine natural products biemamide B and D as potential TGF-β inhibitors11,12. Secondly, the diastereoselective synthesis of β-(aziridin-2-yl)-β-hydroxy ketones was achieved by Mukaiyama aldol reaction of optically pure 1-(1-phenylethyl)-aziridine-2-carboxaldehyde and various enol silanes in the presence of ZnCl2, in high yield (>82%) with almost perfect stereoselectivity (98:2 dr) via a chelation-controlled transition state. These were used for the asymmetric synthesis of epiallo-isomuscarine alkaloids13,14,15.
Here, we report the synthesis of enantiopure aziridine-2-carboxylates. The diastereomeric mixture of (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate (2) and (S)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate (3) (4.1 g, 90%) were prepared in quantitative yield from 2,3 -dibromopropane (-)-mentholyl ester and (1R)-phenylethylamine as shown in Figure 2. The successful selective crystallization method gave rise to enantiopure forms of each isomer, (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate (2) from MeOH and (S)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate (3) from n-pentane in Figure 3. This protocol is applicable for manufacturing several hundred kilograms of enantiopure aziridine-2-carboxylates. Transesterification reactions of either 2 or 3 using either Mg or potassium carbonate with alcohols such as MeOH and EtOH yielded corresponding alkyl ester of chiral aziridine-2-carboxylates. Chiral aziridine ethyl ester (3) was also prepared easily from the reaction of 2,3-dibromopropane ethyl ester and (1R)-phenylethylamine followed by flash column chromatography. This procedure is performed easily in the laboratory to get a few grams of the corresponding chiral aziridine-2-carboxylates.
We achieved nucleophilic ring-opening reaction of chiral ethyl 1-((R)-1-phenylethyl)aziridine-(2S)-carboxylate with N3 yielded ethyl (2R)-azido-3-(((R)-1-phenylethyl)amino)propanoate in a regioselective and stereoselective manner10. This enantiopure azido propionate (5) was cyclized to 5-amino-hexahydro-pyrimidine-2,4-dione (6) which was converted for the synthesis of common structural core (9H-Fluoren-9-yl)methyl(3-(((R)-3-methyl-2,4-dioxo-1-((R)-1 phenylethyl)hexahydro pyrimidin-5-yl)amino)-3-oxopropyl)carbamate (7) of biemamide B and D shown in Figure 4. From this core moiety 7, the synthesis of 5,6-dihydrouracil containing biemamides B (8) and D (9), which are potential TGF-β inhibitors, could be achieved as shown in Figure 4.
We performed stereoselective Mukaiyama aldol reaction of chiral aziridine-2-carboxaldehyde (10) with silyl enol ether 11 to yield β-(aziridin-2-yl)-β-hydroxy ketone12 in high yield (85%) with outstanding diastereoselectivity (>98:2). The selective reduction of compound 13 as a TBS-protected form of 12 with L-selectride (1.1 equiv.) at -78 °C gave rise to unexpected tetrahydrofuran 15 in 15% yield along with the expected alcohol 14. The same reaction with twice the amount of L-selectride (2.0 equiv.) at -78 °C followed by stirring at room temperature for 8 h, yielded 15 in 95% yield along with the reduced hydroxy compound 14 in 5% yield. This compound 15 was derived from the regioselective ring-opening of aziridine by an internal hydroxy group as a nucleophile in 14. Utilization of this compound 15 provides a new route for the synthesis of (-)-epiallo-isomuscarine (17) in an efficient manner (Figure 5).
For characterization of all the compounds, 1H and 13C NMR spectroscopy and mass spectrometry (HRMS-ESI) were used to confirm the structure and assess the purity of the products. Key data for representative compounds are described in Supplementary File 1.
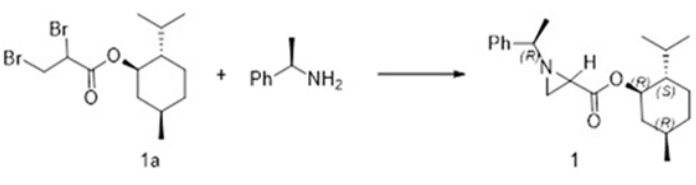
Figure 2: Preparation of (-)-mentholyl (1R)-phenylethyl-(2R)-(2) and (2S)-aziridine-2- carboxylate 2' from 2,3-dibromopropane (-)-mentholyl ester (1a)7,13. Please click here to view a larger version of this figure.
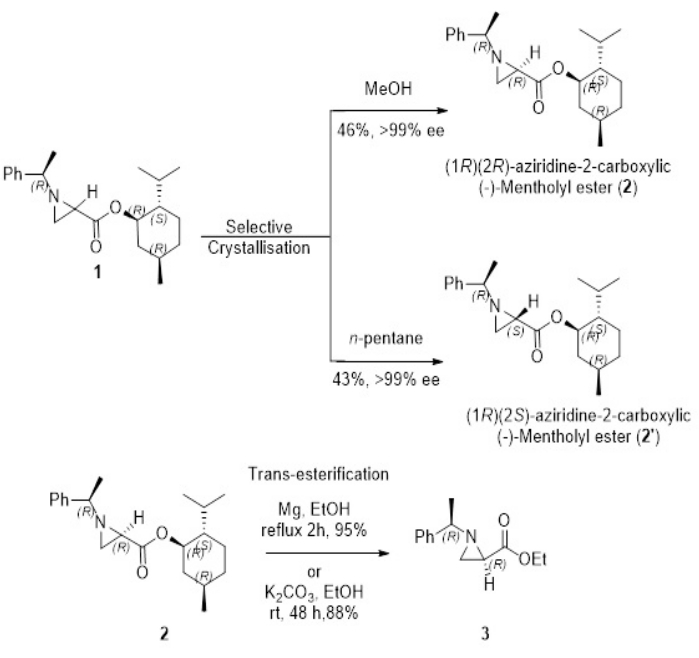
Figure 3: Preparation of both enantiomer of (-)-mentholyl (2R)- and (2S)-aziridine-2- carboxylate, and ethyl (2R)-aziridine-2-carboxylate via trans-esterification (3) as a representative example7,13. Please click here to view a larger version of this figure.
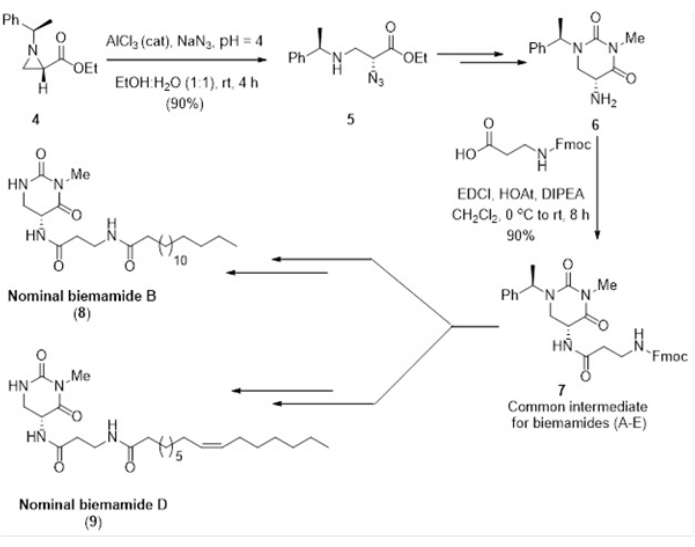
Figure 4: Regioselective ring opening of ethyl (2S)-aziridine-2-carboxylate (4) via azide nucleophile followed by sequential reactions for the total synthesis of biemamide B (8) and biemamide D (9)11. This figure has been modified with permission from Srivastava et al.11. Please click here to view a larger version of this figure.
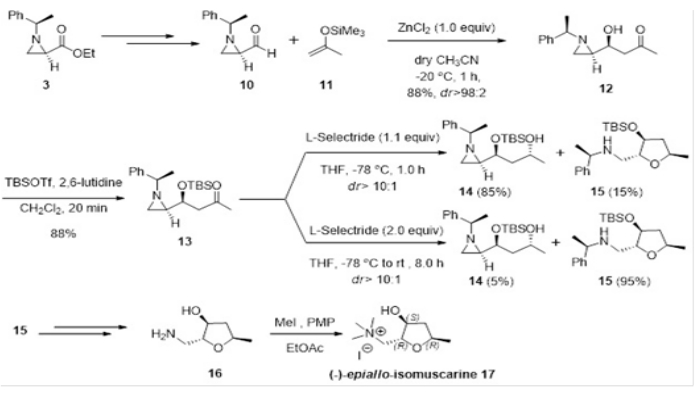
Figure 5: Stereoselective Mukaiyama aldol reaction of chiral aziridine-2-carboxaldehyde (10) and silyl enol ether (11) yielding an adduct 12 and its regio- and stereoselective transformations for the total synthesis of (-)-epiallo-isomuscarine (17)13. Please click here to view a larger version of this figure.
Refer to Figures 6 para 16 for 1H and 13C NMR spectra of representative compounds7,8,11.
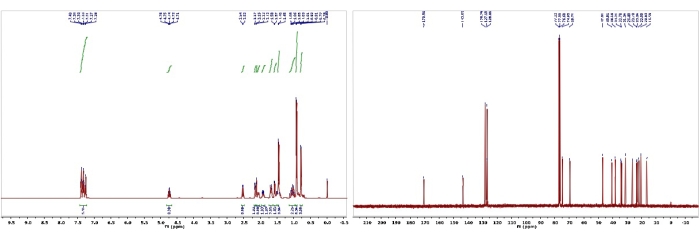
Figure 6: 1H and 13C NMR spectra of 2. Chemical shifts and relative integrations of characteristic protons and carbons are shown13. Please click here to view a larger version of this figure.
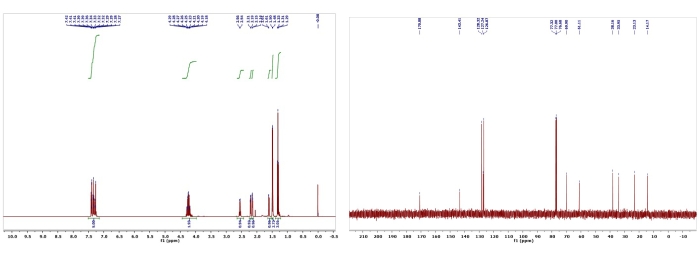
Figure 7: 1H and 13C NMR spectra of 3. Chemical shifts and relative integrations of characteristic protons and carbons are shown13. Please click here to view a larger version of this figure.
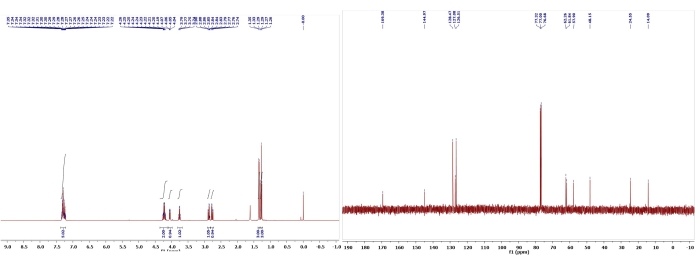
Figure 8: 1H and 13C NMR spectra of 5. Chemical shifts and relative integrations of characteristic protons and carbons are shown11. This figure has been modified with permission from Srivastava et al.11. Please click here to view a larger version of this figure.
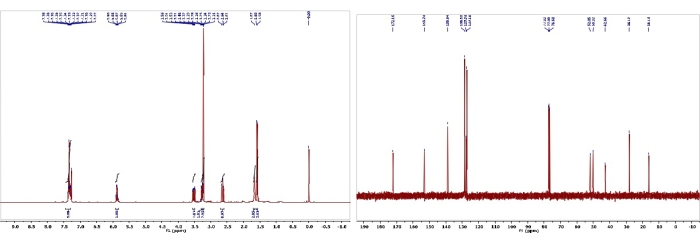
Figure 9: 1H and 13C NMR spectra of 6. Chemical shifts and relative integrations of characteristic protons and carbons are shown11. This figure has been modified with permission from Srivastava et al.11. Please click here to view a larger version of this figure.
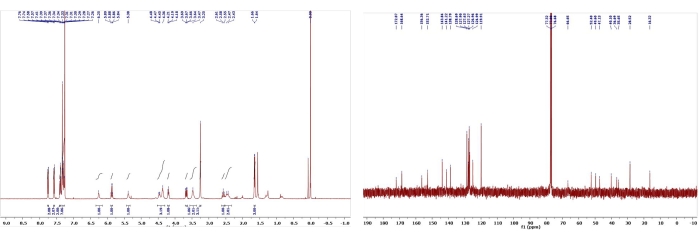
Figure 10: 1H and 13C NMR spectra of 7. Chemical shifts and relative integrations of characteristic protons and carbons are shown11. This figure has been modified with permission from Srivastava et al.11. Please click here to view a larger version of this figure.
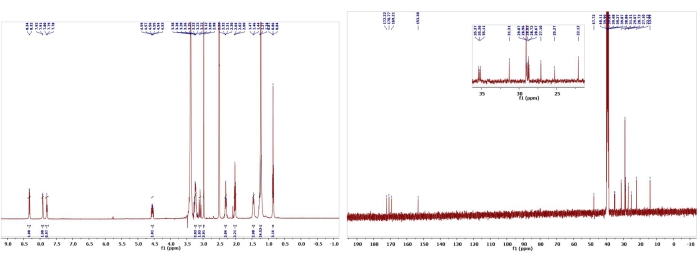
Figure 11: 1H and 13C NMR spectra of Biemamide B (8). Chemical shifts and relative integrations of characteristic protons and carbons are shown11. This figure has been modified with permission from Srivastava et al.11. Please click here to view a larger version of this figure.
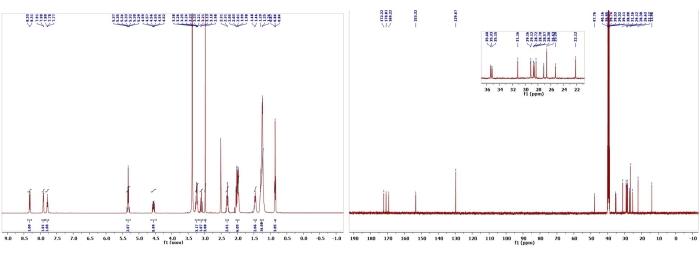
Figure 12: 1H and 13C NMR spectra of Biemamide D (9). Chemical shifts and relative integrations of characteristic protons and carbons are shown11. This figure has been modified with permission from Srivastava et al.11. Please click here to view a larger version of this figure.
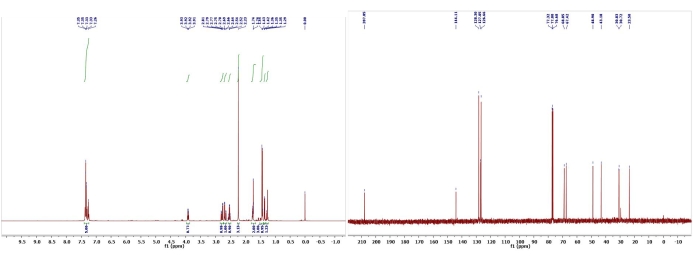
Figure 13: 1H and 13C NMR spectra of 12. Chemical shifts and relative integrations of characteristic protons and carbons are shown13. This figure has been modified with permission from Srivastava et al.13. Please click here to view a larger version of this figure.
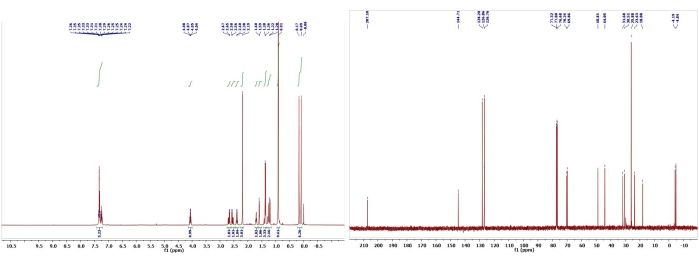
Figure 14: 1H and 13C NMR spectra of 13. Chemical shifts and relative integrations of characteristic protons and carbons are shown13. This figure has been modified with permission from Srivastava et al.13. Please click here to view a larger version of this figure.
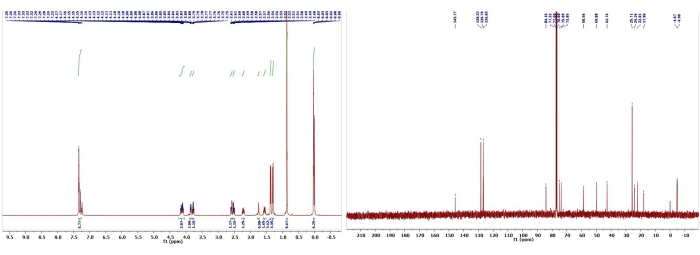
Figure 15: 1H and 13C NMR spectra of 15. Chemical shifts and relative integrations of characteristic protons and carbons are shown13. This figure has been modified with permission from Srivastava et al.13. Please click here to view a larger version of this figure.
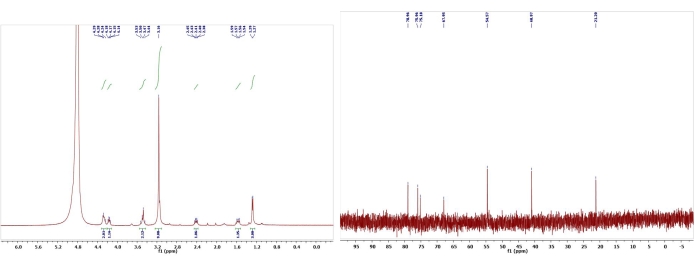
Figure 16: 1H and 13C NMR spectra of (-)-epiallo-isomuscarine (17). Chemical shifts and relative integrations of characteristic protons and carbons are shown13. This figure has been modified with permission from Srivastava et al.13. Please click here to view a larger version of this figure.
Supplementary File 1: Spectral data of all compounds in this manuscript. Please click here to download this File.
